UNG THƯ BIỂU MÔ TẾ BÀO THẬN
BSNT. Nguyễn Minh Hiếu
Đại cương
Ung thư thận là loại ung thư thường gặp đứng thứ 14 trên toàn cầu, với hơn 430.000 ca mới được chẩn đoán vào năm 2020, và 434840 ca mắc trên toàn cầu vào năm 2022. Tỷ lệ mắc thay đổi theo khu vực địa lý, cao hơn ở châu Âu và Bắc Mỹ. Ung thư biểu mô tế bào thận chiếm khoảng 90% tổng số các ung thư thận.
Trong khi tỷ lệ mắc ung thư thận có xu hướng tăng đều theo thời gian, bao gồm cả sự gia tăng chậm trong thập kỷ vừa qua, thì tỷ lệ tử vong lại giảm dần. Điều này một phần được lý giải bởi sự gia tăng phát hiện tình cờ qua các phương tiện chẩn đoán hình ảnh ổ bụng.
Về mặt điều trị, những tiến bộ trong hiểu biết về cơ sở di truyền, các con đường phân tử của ung thư biểu mô tế bào thận đã dẫn đến sự phát triển và phê duyệt nhiều liệu pháp nhắm trúng đích mới, tạo ra thay đổi lớn trong thực hành lâm sàng. Điều trị ung thư thận khu trú đã chuyển dịch từ phẫu thuật cắt thận tận gốc mở sang các phương pháp ít xâm lấn hơn như cắt thận bán phần, đốt nhiệt và theo dõi chủ động. Đồng thời, điều trị ung thư thận di căn đã có bước tiến rõ rệt, từ một bệnh gần như không thể kiểm soát sang đạt được tỷ lệ đáp ứng và kiểm soát bệnh trên 50%, kèm theo cải thiện đáng kể thời gian sống còn đặc hiệu ung thư.
Với sự hiểu biết ngày càng sâu về tính không đồng nhất sinh học của bệnh và mối liên quan với các liệu pháp kháng tạo mạch và miễn dịch, ung thư thận được kỳ vọng sẽ dần trở thành một bệnh mạn tính trong tương lai.
Nguyên nhân, các yếu tố nguy cơ
Nguyên nhân gây bệnh của ung thư biểu mô tế bào thận vẫn chưa được xác định rõ. Tuy nhiên, có nhiều yếu tố nguy cơ liên quan đến bệnh đã được tìm ra, trong đó có những yếu tố liên quan đến di truyền (không thể dự phòng) và những yếu tố không liên quan đến di truyền (có thể dự phòng được).
Những yếu tố không liên quan đến di truyền
Có nhiều yếu tố nguy cơ đã được xác lập đối với ung thư biểu mô tế bào thận như hút thuốc lá, béo phì, tăng huyết áp và phơi nhiễm hóa chất.
Những yếu tố liên quan đến di truyền
Ước tính khoảng 6%–9% các ung thư thận có đột biến dòng mầm ở các gen liên quan đến khuynh hướng ung thư. Nhiều hội chứng di truyền trội trên nhiễm sắc thể thường đã được mô tả, bao gồm hội chứng von Hippel–Lindau, hội chứng u cơ trơn di truyền và ung thư biểu mô tế bào thận hoặc ung thư biểu mô tế bào thận thiếu hụt fumarate hydratase, ung thư biểu mô tế bào thận dạng nhú di truyền, phức hợp xơ cứng củ, hội chứng Birt–Hogg–Du bé và ung thư biểu mô tế bào thận thiếu hụt succinate dehydrogenase.
Triệu chứng lâm sàng
Triệu chứng kinh điển
Các triệu chứng lâm sàng kinh điển của ung thư biểu mô tế bào thận gồm: Đau hông lưng (40%); sờ thấy khối vùng bụng (30-40%);tiểu máu (60%). Tam chứng kinh điển chỉ gặp ở dưới 10% bệnh nhân ung thư biểu mô tế bào thận mới được chẩn đoán. Do khoang sau phúc mạc có thể chứa đựng sự phát triển đáng kể của khối u trước khi xuất hiện triệu chứng, chỉ các khối ung thư thận kích thước lớn mới có thể phát hiện bằng sờ nắn.
Hiện nay, việc sử dụng rộng rãi các phương tiện chẩn đoán hình ảnh ổ bụng dẫn đến phát hiện ung thư biểu mô tế bào thận tình cờ trong khoảng 37% đến 61% trường hợp. Cùng với sự gia tăng phát hiện tình cờ, tiểu máu đại thể hiện chỉ được ghi nhận ở dưới 25% bệnh nhân và thường gặp hơn ở giai đoạn bệnh tiến triển. Khoảng 1,3% bệnh nhân có tiểu máu đại thể được chẩn đoán là ung thư biểu mô tế bào thận.
Hội chứng cận u
Các hội chứng cận u xuất hiện ở khoảng 10% đến 40% bệnh nhân ung thư biểu mô tế bào thận và giữa các nghiên cứu không ghi nhận mối liên quan nhất quán với giai đoạn hoặc độ mô học cao hơn. Các biểu hiện cận u thường gặp bao gồm sốt (8%), tăng calci máu (1%–30%), thiếu máu (22%–52%), tăng tiểu cầu (8%–12%), tăng hồng cầu (2%–4%) và tăng huyết áp (3%–18%).
Tăng hồng cầu cận u liên quan đến nồng độ erythropoietin tăng cao, được sản xuất bởi tế bào u ung thư biểu mô tế bào thận khi VHL bị bất hoạt.
Hội chứng Stauffer, một hội chứng cận u được mô tả lần đầu năm 1961, đặc trưng bởi tăng men gan ở khoảng 3% bệnh nhân ung thư biểu mô tế bào thận; gan lách to không kèm di căn gan cũng có thể gặp trong hội chứng này.
Các hội chứng cận u có thể thoái lui ở tới 52%bệnh nhân sau cắt thận hoặc điều trị toàn thân ung thư biểu mô tế bào thận, và sự tồn tại dai dẳng của các triệu chứng cận u sau cắt thận có thể gợi ý bệnh tồn dư.
Cận lâm sàng
Giải phẫu bệnh
Bằng chứng về mô bệnh học là tiêu chuẩn bắt buộc để chẩn đoán ung thư biểu mô tế bào thận. Nhưng không phải mọi trường hợp phát hiện khối u đặc tại thận đều cần phải sinh thiết.
Chỉ định sinh thiết thận
Sinh thiết khối u thận không phải là xét nghiệm thường quy cho mọi khối u tại thận (renal mass), mà chỉ được thực hiện khi kết quả mô học có khả năng thay đổi chiến lược điều trị, và khi lợi ích vượt trội nguy cơ, đồng thời bệnh nhân và ê-kíp điều trị đồng thuận. Đây là cách tiếp cận hiện đại, cá thể hóa, tránh sinh thiết không cần thiết nhưng cũng tránh bỏ sót những tình huống cần chẩn đoán mô học trước điều trị.
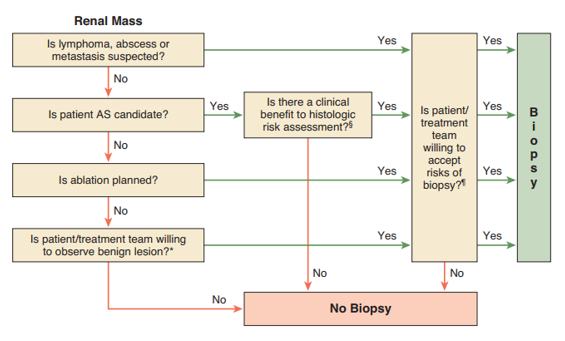
Hình 1. Lưu đồ hướng dẫn chỉ định sinh thiết thận
(Nguồn: Kutikov A, Smaldone MC, Uzzo RG, Haifler M, Bratslavsky G, LeibovichBC. Renal Mass Biopsy: Always, Sometimes, or Never? Eur Urol2016;70(3):403–406.)
Chú giải:
Trước hết, khi phát hiện khối tổn thương đặc tại thận (renal mass), bước đầu tiên là đặt câu hỏi có nghi ngờ lymphoma, áp xe thận hoặc di căn hay không. Nếu có, sinh thiết gần như luôn được chỉ định, vì các tình huống này không được điều trị bằng phẫu thuật cắt thận tiêu chuẩn mà cần chẩn đoán mô học để định hướng điều trị nội khoa hoặc điều trị toàn thân. Nếu không nghi ngờ, chuyển sang nhánh tiếp theo.
Bước thứ hai là đánh giá bệnh nhân có phải là ứng viên của active surveillance (theo dõi tích cực) hay không. Đây thường là bệnh nhân lớn tuổi, nhiều bệnh nền, khối u nhỏ, tăng trưởng chậm, tiên lượng sống hạn chế. Nếu có, câu hỏi tiếp theo không phải là sinh thiết ngay mà là: việc đánh giá nguy cơ mô học (histologic risk assessment) có mang lại lợi ích lâm sàng hay không. Nghĩa là: kết quả sinh thiết có thực sự làm thay đổi quyết định điều trị hay không. Nếu có lợi ích, tiếp tục cân nhắc sinh thiết; nếu không, thì không sinh thiết.
Nếu bệnh nhân không phải ứng viên theo dõi tích cực, bước tiếp theo là xem có kế hoạch điều trị đốt u tại chỗ (ablation) hay không, ví dụ đốt u bằng sóng cao tần hoặc vi sóng. Trong trường hợp có kế hoạch ablation, sinh thiết thường được khuyến cáo trước hoặc trong quá trình can thiệp, vì ablation phá hủy mô và sẽ không còn mô để chẩn đoán về sau. Do đó, nhánh này thường dẫn tới sinh thiết, miễn là bệnh nhân và ê-kíp điều trị chấp nhận nguy cơ.
Nếu không có kế hoạch ablation, sơ đồ tiếp tục hỏi: bệnh nhân hoặc ê-kíp điều trị có sẵn sàng chỉ theo dõi một tổn thương được cho là lành tính hay không. Ví dụ: u mỡ mạch (angiomyolipoma) điển hình, oncocytoma nghi ngờ cao trên hình ảnh. Nếu có thể chấp nhận theo dõi, thì không sinh thiết. Nếu không chấp nhận, nghĩa là vẫn cần xác định bản chất mô học, thì tiếp tục cân nhắc sinh thiết.
Tất cả các nhánh “có thể sinh thiết” đều hội tụ vào một câu hỏi then chốt cuối cùng ở cột bên phải: bệnh nhân và ê-kíp điều trị có chấp nhận các nguy cơ của sinh thiết hay không. Các nguy cơ này bao gồm chảy máu, nhiễm trùng, sinh thiết không chẩn đoán được, hoặc rất hiếm là gieo rắc tế bào u theo đường kim. Nếu chấp nhận, thì tiến hành sinh thiết. Nếu không chấp nhận, dù về mặt lý thuyết có chỉ định, thì quyết định cuối cùng vẫn là không sinh thiết.
Phân loại RCC theo giải phẫu bệnh
Phân loại giải phẫu bệnh đóng vai trò quan trọng trong chẩn đoán, tiên lượng và điều trị ung thư biểu mô tế bào thận (RCC). Hầu hết các hệ thống phân loại đều dựa trên hình thái học và mô bệnh học của khối u, tuy nhiên, ngày càng có xu hướng tích hợp các đặc điểm di truyền. Theo hệ thống phân loại hiện tại của Tổ chức Ytế Thế giới (WHO), có 16 loại phụ của RCC, trong đó bốn biến thể chính bao gồm ung thư biểu mô tế bào sáng (clear cell RCC - ccRCC), ung thư biểu mô nhú (papillary RCC), ung thư biểu mô kỵ sắc (chromophobe RCC) và ung thư biểu mô ống góp (collecting duct carcinoma - CDC). Nhóm này chiếm khoảng 90 - 95% tổng số trường hợp RCC, ngoài ra còn có các loại ít gặp hơn và một nhóm không phân loại.Một số phân nhóm hiếm được bổ sung trong hệ thống phân loại WHO bao gồm ung thư biểu mô ống nang, ung thư biểu mô tế bào sáng/papillary hỗn hợp, ung thư biểu mô liên quan đến bệnh nang thận, ung thư biểu mô liên quan đến hội chứng leiomyomatosis di truyền (HLRCC), và ung thư biểu mô tế bào thận liên quan đến thiếu hụt succinate dehydrogenase (SDH). Xu hướng chuyển từ phân loại dựa trên hình thái học sang phân loại phân tử trong RCC phản ánh sự tiến bộ trong hiểu biết về cơ chế bệnh sinh và có thể dần thay thế các hệ thống phân loại truyền thống.
Bảng 1. Phân loại ung thư biểu mô tế bào thận rải rác theo Tổ chức Y tế Thế giới WHO 2004, kèm theo đặc điểm di truyền và lâm sàng
Loại | Đặc điểm di truyền | Đặc điểm lâm sàng |
Ung thư biểu mô tế bào thận tế bào sáng (Clear cell RCC) (70%–80%) | Bệnh Von Hippel–Lindau (VHL) là thể gia đình. Mất đoạn, đột biến hoặc methyl hóa vùng 3p25–26 (gen VHL). | Thể thường gặp nhất. Tiên lượng phụ thuộc vào giai đoạn và độ mô học. Đáp ứng với thuốc ức chế tyrosine kinase và thuốc ức chế PD-1/PD-L1. |
Ung thư biểu mô tế bào thận nhú type 1 (5%–10%) | Ung thư biểu mô tế bào thận nhú di truyền (HPRCC) là thể gia đình. Đột biến vùng 7q31 (gen MET) trong HPRCC và trong 81% các trường hợp rải rác. Tăng số lượng NST 7 hoặc 17 (tam bội hoặc tứ bội), mất đoạn 9p. | Thường đa ổ. Tỷ lệ sống thêm đặc hiệu ung thư sau 5 năm >95%. Đáp ứng với thuốc ức chế tyrosine kinase kém hơn. |
Ung thư biểu mô tế bào thận nhú type 2 (5%–10%) | Tăng số lượng NST 7 hoặc 17 (tam bội hoặc tứ bội), mất đoạn 9p. | Tiên lượng xấu hơn papillary RCC type 1; tương đương với clear cell RCC. Có nguồn gốc từ ống lượn gần. |
Ung thư biểu mô tế bào thận tế bào kỵ sắc (Chromophobe RCC) (3%–5%) | Mất nhiễm sắc thể diện rộng gồm Y, 1, 2, 6, 10, 13, 17, 21. Đột biến vùng 17p11.2 khi liên quan đến hội chứng Birt–Hogg–Dubé (BHD). | Chiếm khoảng 5% các RCC. Gặp ở nam và nữ như nhau, tiên lượng chung rất tốt. |
Ung thư biểu mô tế bào thận không phân loại (1%–3%) | Đa dạng. | Tiên lượng nhìn chung kém. |
Ung thư biểu mô tế bào thận liên quan bệnh thận nang mắc phải (hiếm) | Chưa xác định. | Tiên lượng rất tốt. |
Ung thư biểu mô ống góp (u Bellini) (hiếm) | Rất biến thiên. Mất đoạn 1q, 6p, 8p, 9p, 13q, 19q, 21q. | Nam gặp nhiều hơn nữ (2:1). Tuổi trung bình 55. Vi thể độ ác tính cao, có thể giống phổ ung thư biểu mô niệu mạc. Tiên lượng chung xấu. |
Ung thư biểu mô tế bào thận liên quan hội chứng u cơ trơn di truyền và RCC (HLRCC) (hiếm) | Đột biến gen fumarate hydratase (FH) tại vùng 1q42–43. | Chỉ xuất hiện trong bối cảnh HLRCC gia đình. Tiên lượng thường xấu, do đó cần điều trị sớm (ngoại lệ của quy tắc 3 cm). |
Ung thư biểu mô tế bào thận do chuyển đoạn họ MiT (hiếm) | Các đột biến khác nhau liên quan đến NST Xp11.2 gây dung hợp gen TFE3. | Gặp ở trẻ em và người trẻ. Có thể phát hiện ở giai đoạn muộn và diễn tiến ác tính hơn ở người lớn. |
Ung thư biểu mô tế bào thận dạng ống nhầy và tế bào hình thoi (hiếm) | Chưa xác định. | Nữ gặp nhiều hơn nam (4:1). Hiếm khi di căn. |
Tân sinh thận dạng nang đa ổ tế bào sáng, tiềm năng ác tính thấp (1%–5%) | Giống hệt clear cell RCC. | Biến thể của ccRCC. Đặc điểm lâm sàng gần như luôn lành tính. |
Ung thư biểu mô tế bào thận sau neuroblastoma (hiếm) | Chưa xác định. | Chỉ gặp ở trẻ em có tiền sử neuroblastoma. Hình thái đại thể và vi thể tương tự clear cell RCC. |
Ung thư biểu mô tủy thận (hiếm) | Chưa xác định. | Liên quan đến người mang đặc điểm hồng cầu hình liềm. Diễn tiến rất ác tính và gây tử vong trong vòng 12 tháng. Tuổi trung bình 19. Nam > nữ. |
Ung thư biểu mô thận thiếu hụt succinate dehydrogenase (hiếm) | Các tiểu đơn vị của phức hợp succinate dehydrogenase: SDHB (1p36.1–35) hoặc SDHD (11q23). | Chưa xác định. |
Ung thư biểu mô tế bào thận dạng ống – nang (Tubulocystic RCC) (hiếm) | Chưa xác định. | Tiên lượng tốt. |
Phân độ mô học
Ngoài đặc điểm mô học, các yếu tố khác như độ mô học nhân, biệt hóa dạng sarcomatoid/rhabdoid, hoại tử khối u và xâm lấn mạch máu cũng có ý nghĩa quan trọng trong tiên lượng bệnh. Hệ thống phân loại Fuhrman trước đây được sử dụng phổ biến để đánh giá độ mô học trong ccRCC, nhưng giá trị tiên lượng của nó đối với các loại RCC khác vẫn chưa được xác định rõ ràng. Do đó, WHO đã giới thiệu hệ thống phân loại WHO/ISUP với bốn bậc để đánh giá ccRCC và papillary RCC, tuy nhiên, hệ thống này chưa được áp dụng cho chromophobe RCC. Trong các yếu tố tiên lượng, biệt hóa sarcomatoid xuất hiện ở khoảng 5% trường hợp RCC và có thể gặp ở bất kỳ phân nhóm nào, thường liên quan đến tiên lượng xấu. Hoại tử vi thể hoặc đại thể có tương quan với tiên lượng trong ccRCC, trong khi mối liên hệ giữa xâm lấn vi mạch hoặc đại mạch với tiên lượng bệnh vẫn chưa được xác định rõ ràng.
Bảng 2. Hệ thống phân độ mô học của Tổ chức Y tế Thế giới và Hiệp hội Giải phẫu bệnh Tiết niệu Quốc tế (ISUP) cho ung thư biểu mô tế bào thận tế bào sáng và dạng nhú
Đặc điểm giải phẫu bệnh của một số thể RCC phổ biến
Ung thư biểu mô tế bào thận tế bào sáng (70-80%)
Đại thể
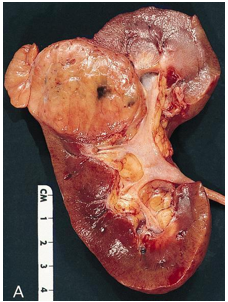
Thường là khối u vỏ thận một bên và đơn ổ, kích thước trung bình khoảng 7 cm. Khối u thường có ranh giới rõ, được bao quanh bởi giả bao u, phát triển theo kiểu đẩy nở và lồi ra từ vỏ thận. Hình thái đại thể đa dạng với thành phần đặc và nang, kèm các vùng xơ hóa màu xám và các ổ xuất huyết mới hoặc cũ màu nâu; hoại tử và biến đổi nang gặp thường xuyên. Khối u có màu vàng ánh kim do hàm lượng lipid cao; các khối u có độ mô học cao hơn có thể không có màu vàng do hàm lượng lipid và glycogen thấp hơn. Các vùng mềm, dạng thịt có thể phản ánh sự hiện diện của biệt hóa dạng sarcomatoid. Thường xuyên ghi nhận xâm lấn tĩnh mạch thận và xoang thận. Tổn thương hai bên và đa ổ là đặc điểm gợi ý bệnh lý di truyền.
Về phân giai đoạn, các khối u có kích thước lớn hơn 7 cm hầu như luôn xâm lấn mỡ xoang thận; nếu không quan sát thấy xâm lấn ở các khối u lớn, cần tiến hành xem xét lại đại thể bổ sung. Xâm lấn bao thận được đặc trưng bởi sự tiến triển không đều và gián đoạn của khối u vào mô mỡ quanh thận kèm theo mất đường viền ngoài lồi đều, nhẵn của thận; khối u lồi đều, nhẵn và được bao phủ bởi giả bao ung thư không được xem là xâm lấn mỡ quanh thận. Để chẩn đoán xâm lấn, tế bào u phải tiếp xúc trực tiếp với mô mỡ hoặc xâm nhập dạng các lưỡi không đều vào mô quanh thận, có hoặc không kèm phản ứng xơ sinh. Xâm lấn xoang thận là con đường lan tràn ngoài thận thường gặp nhất và thường xảy ra trước xâm lấn bao thận; khác với bao thận,xoang thận không được ngăn cách với nhu mô thận bằng một bao xơ riêng biệt. Không được coi là xâm lấn thực sự nếu khối u vẫn được ngăn cách với các cấu trúc xoang bởi một viền nhu mô thận; được coi là xâm lấn xoang thận khi khối u lồi rõ vào mô mỡ xoang thận vượt quá nhu mô thận, kể cả khi vẫn được bao phủ bởi mô liên kết thưa. Việc khối u bao quanh các cấu trúc mạch máu hoặc bạch mạch lớn là dấu hiệu gợi ý xâm lấn mỡ xoang thận. Xâm lấn mạch máu có thể biểu hiện dưới dạng các nốt u trong xoang thận; trong trường hợp lòng mạch bị bít một phần, sự hiện diện của một lớp nội mô đơn độc phủ lên bề mặt khối u không loại trừ xâm lấn mạch máu. Xâm lấn tĩnh mạch nhỏ thường hàm ý xâm lấn tĩnh mạch lớn, và xâm lấn xoang thận thường đồng nghĩa với xâm lấn tĩnh mạch thận, do đó cần được thăm khám và đánh giá một cách thận trọng.
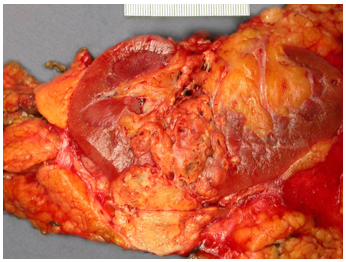
Hình 2. ccRCC đã xâm lấn mạc Gerotta (pT4)
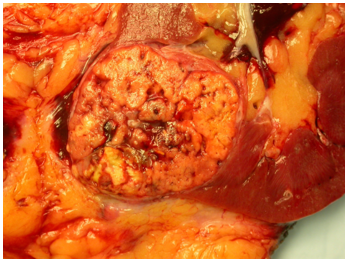
Hình 3. ccRCC có màu vàng, giới hạn ở thận (pT1b)
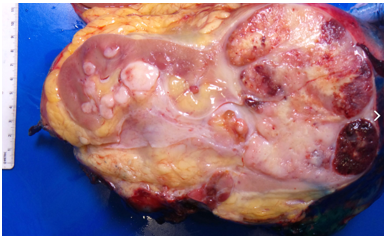
Hình 4. ccRCC đa ổ, với diện cắt đại thể loang lổ,khối u màu vàng óng xen kẽ các vùng xơ hóa và xuất huyết. Các vùng mềm, dạng thịt, màu nâu nhạt đến trắng xám trong khối u có thể gợi ý sự biệt hóa sarcomatoid.
Vi thể
Thường gặp các đám và bè tế bào sắp xếp chặt chẽ, tế bào có bào tương sáng và màng tế bào rõ. Bào tương hạt bắt màu eosin có thể được quan sát trong các khối u độ mô học cao hoặc ở vùng lân cận các ổ xuất huyết hay hoại tử. Hệ thống các mạch máu nhỏ, thành mỏng,phân nhánh dạng cây là một đặc điểm chẩn đoán quan trọng, đặc biệt trong các trường hợp có bào tương hạt eosin. Các kiểu cấu trúc kiến trúc gồm dạng đặc, dạng phế nang hay dạng ổ (alveolar hoặc nested), dạng tuyến nang hay ống (acinar hoặc tubular), dạng vi nang chứa hồng cầu thoát mạch hoặc dịch ái toan, và đôi khi dạng đại nang. Có thể gặp cấu trúc nhú khu trú, tuy nhiên sự hình thành nhú nổi bật làm tăng khả năng các phân nhóm khác như u thận tế bào sáng dạng nhú, ung thư biểu mô tế bào thận có tái sắp xếp TFE3, biến đổi TFEB hoặc đột biến ELOC. Mô đệm thường không đặc hiệu, không có phản ứng xơ sinh, trái ngược với ung thư biểu mô ống góp hoặc ung thư biểu mô niệu mạc, và chỉ có đáp ứng viêm tối thiểu. Các đặc điểm độ mô học cao bao gồm biệt hóa dạng rhabdoid với các tế bào ác tính lớn,độ ác tính cao, bào tương eosin đồng nhất phong phú, nhân lệch tâm và các thể vùi eosin hình cầu trong bào tương, cũng như biệt hóa dạng sarcomatoid, có thể xảy ra ở bất kỳ phân nhóm ung thư biểu mô tế bào thận nào. Hoại tử khối u là một đặc điểm có thể gặp. Các biến thể mô học ít gặp, hiện chưa rõ ý nghĩa tiên lượng,bao gồm dạng nang, dạng giả nhú, tạo xương lạc chỗ, các thể cầu hyaline trong và ngoài tế bào, các thể vùi bào tương bắt màu kiềm, sự hiện diện phong phú của tế bào khổng lồ đa nhân, các u hạt dạng sarcoid hoặc hình ảnh myospherulosis. Ung thư biểu mô tế bào thận tế bào sáng có đột biến BAP1 thường biểu hiện cấu trúc nhú, bào tương eosin và các thể cầu trong bào tương. Trên thực hành, các vùng u độ mô học thấp hơn với hình thái điển hình của ung thư biểu mô tế bào thận tế bào sáng là những vùng hữu ích nhất cho chẩn đoán, trong khi các khối u độ mô học cao hơn có thể biểu hiện các đặc điểm chồng lấp với các тип ung thư biểu mô tế bào thận khác.
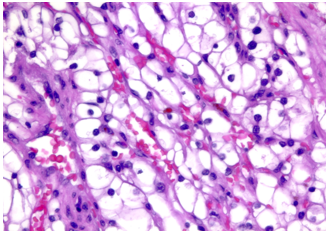
Hình 5. ccRCC với hệ thống mạch xoang dạng cây phân nhánh, thành mạch mỏng
Hình 6. ccRCC, có biến đổi vi nang và đại nang, kèm hiện tượng thoát hồng cầu
Ung thư biểu mô tế bào thận thể nhú (10-20%)
Đại thể
Đa số khối u có ranh giới rõ, được bao quanh bởi giả bao u. Mặt cắt đại thể có màu vàng nâu, nâu đỏ hoặc đa dạng. Bề mặt cắt có thể có tính chất hạt hoặc dễ vỡ, phù hợp với hình thái dạng nhú. Các vùng hoại tử và thoái hóa dạng nang xuất hiện với mức độ thay đổi.
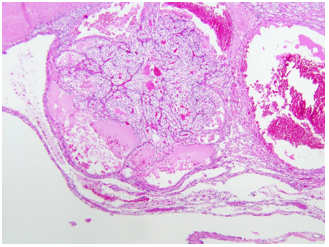
Hình 7. pRCC diện cắt đại thể dạng hạt, biểu hiện các nhú u, kèm hoại tử và xuất huyết.
Vi thể
Kiến trúc chủ yếu là dạng nhú hoặc dạng ống–nhú. Lớp lót gồm các tế bào có hình thái đa dạng, từ các tế bào vuông nhỏ bắt màu kiềm với hạch nhân không rõ cho đến các tế bào lớn hơn,bào tương eosin với hạch nhân nổi bật; có thể gặp bào tương sáng, đây là một bẫy chẩn đoán đã biết trong ung thư biểu mô tế bào thận dạng nhú. Các tế bào có thể sắp xếp theo kiểu hàng thẳng hoặc giả tầng. Tính không đồng nhất và hình thái hỗn hợp rất thường gặp, chiếm khoảng 47–48% các trường hợp, do đó đây là một trong những lý do khiến việc phân typ không còn được ưu tiên. Thường thấy thâm nhiễm các đại thực bào bọt. Có thể hiện diện các thể cát vôi (psammoma bodies) và sắc tố hemosiderin. Các kiểu hình đã được mô tả gồm: dạng PRCC hai pha (biphasic,phế nang hoặc squamoid), với hai quần thể tế bào, các ổ tế bào lớn hơn dạng squamoid, bào tương eosin, được bao quanh bởi các tế bào nhỏ hơn bắt màu amphophilic tạo thành cấu trúc phế nang; đã ghi nhận bằng chứng diễn tiến ác tính ở tối đa khoảng 15% trường hợp. Dạng PRCC kiểu Warthin, với các nhú bào tương eosin kèm thâm nhiễm lympho dày đặc, tương tự u Warthin của tuyến nước bọt;thường là các khối u độ mô học cao và có thể biểu hiện diễn tiến lâm sàng ác tính. Dạng PRCC đặc hoặc giả đặc, với kiến trúc đặc do các cấu trúc ống và nhú bị nén ép, được lót bởi các tế bào nhỏ có nhân độ thấp; được ghi nhận có diễn tiến lâm sàng tương đối lành tính. Các kiểu kiến trúc mô học liên quan đến tiên lượng xấu hơn bao gồm dạng đặc, dạng vi nhú, hiện tượng hobnailing và kiến trúc vi nang. Hệ thống phân độ WHO/ISUP đã được xác nhận là một yếu tố tiên lượng đối với PRCC, dựa trên mức độ nổi bật của hạch nhân để phân độ 1 đến 3, và dựa trên đa hình nhân tế bào hoặc biệt hóa dạng sarcomatoid hay rhabdoid cho độ 4.
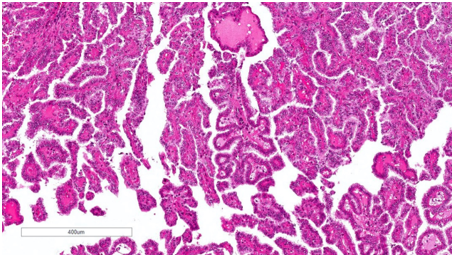
Hình 8. pRCC đặc trưng bởi cấu trúc chủ yếu dạng nhú hoặc ống‑nhú
Ung thư biểu mô tế bào thận tế bào kỵ sắc (3-5%)
Đại thể
Khối u thường có kích thước lớn, với kích thước trung vị khoảng 8 cm và dao động từ 1 đến 30 cm, chủ yếu là dạng đặc, chỉ hiếm gặp các trường hợp dạng nang. Khối u có ranh giới rõ nhưng không có bao thực sự. Mặt cắt có màu nâu nhạt đến xám, trong đó mức độ đậm nhạt thay đổi của màu nâu gụ tương quan với số lượng tế bào có bào tương eosin. Các vùng màu trắng ngà, mềm, dạng thịt gợi ý sự chuyển dạng sarcomatoid. Xuất huyết và hoại tử được ghi nhận trong khoảng 25% trường hợp. Xâm lấn tĩnh mạch thận hiếm gặp. Sẹo trung tâm hiện diện trong khoảng 15% các trường hợp.
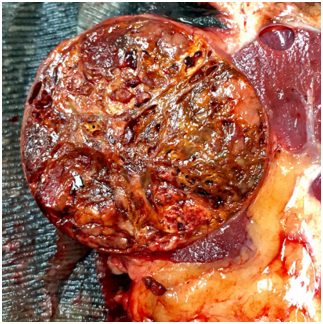
Hình 9. ChRCC, khối u có màu nâu nhạt và tương đối đồng nhất.
Vi thể
Mô tả vi thể (mô học) điển hình cho thấy khối u thường gồm các đám đặc tế bào, được chia cắt bởi các vách xơ–mạch máu không hoàn chỉnh,tạo nên kiểu phát triển bè rộng; kiểu sắp xếp dạng ổ hoặc dạng phế nang gặp íthơn. Các tế bào lớn với bào tương nhạt màu, dạng lưới mịn là đặc trưng nhất và xác định thực thể khối u này, trong đó thuật ngữ chromophobe phản ánh đặc điểm bào tương kém bắt màu. Xen kẽ là các tế bào nhỏ hơn với bào tương eosin. Các tế bào lớn có bào tương rất phong phú, đến mức trong một số mặt cắt, nhân không nằm trong mặt phẳng cắt nên tế bào có vẻ như không nhân, đây là một dấu hiệu gợi ý chẩn đoán mô học mềm. Ranh giới tế bào rõ, tạo hình ảnh giống tế bào thực vật.Có quầng sáng quanh nhân kiểu giả koilocyte. Nhân thường tăng sắc, màng nhân không đều và nhăn nheo, tạo hình ảnh dạng nho khô, kèm hiện tượng hai nhân khá thường gặp.
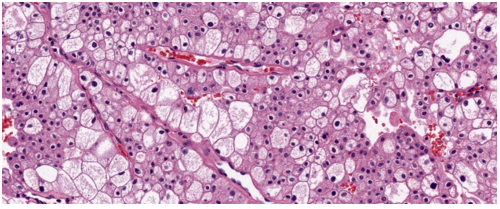
Hình 10. ChRCC thể điển hình được cấu tạo bởi các tế bào đa giác lớn với bào tương sáng, dạng lưới, xen kẽ với các tế bào nhỏ hơn có bào tương eosin. Lưu ý rằng các tế bào lớn có bào tương rất phong phú đến mức trong một số mặt cắt mô, nhân không nằm trong mặt phẳng cắt,khiến các tế bào này có hình ảnh như không nhân.
Ở kiểu hình eosinophilic, ít nhất 80% khối u phải được cấu tạo bởi các tế bào có bào tương eosin; các tế bào này thường nhỏ hơn so với tế bào chromophobe điển hình. Kiến trúc có thể dạng ổ, dạng phế nang hoặc dạng đám đặc, trong đó cấu trúc dạng ổ gặp thường xuyên hơn ở biến thể eosinophilic so với thể điển hình, còn lại các đặc điểm mô học tương tự thể điển hình.
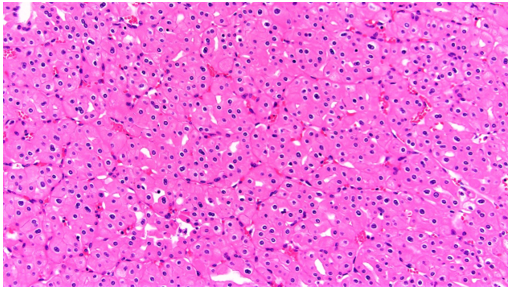
Hình 11. ChRCC kiểu eosinophilic được đặc trưng bởi thành phần chủ yếu, ít nhất 80%, là các tế bào có bào tương eosin, kèm mức độ không điển hình tế bào học nhẹ, với nhân nhăn nheo và hiện tượng hai nhân phân bố rải rác lan tỏa.
Ở dạng sarcomatoid, có hiện tượng mất biệt hóa thành các tế bào hình thoi độ ác tính cao, đa hình, với hoạt động phân bào tăng, bao gồm cả các hình thái phân bào bất thường và hoại tử u.
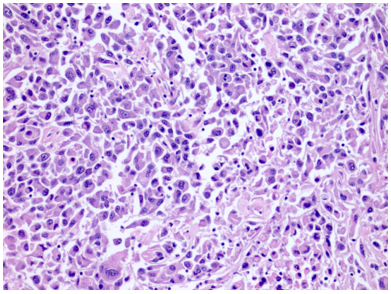
Hình 12. ChRCC kiểu sarcomatoid với các tế bào hình thoi và dạng rhabdoid có mức độ không điển hình cao.
Các kiểu hình khác có thể gặp gồm tế bào khổng lồ đa hình, tế bào dạng rhabdoid, hoặc các tế bào đơn dạng chưa biệt hóa hay anaplastic; tất cả các dạng này có thể kèm theo các thành phần dị dạng mô như dạng tạo xương, dạng hủy xương, tạo sụn, tạo cơ vân hoặc dạng mỡ ác tính. Không có yêu cầu về tỷ lệ tối thiểu của thành phần sarcomatoid để chẩn đoán, tuy nhiên một số tác giả chỉ sử dụng chẩn đoán này khi toàn bộ các vi trường, nếu được xem xét riêng rẽ, có thể bị nhầm lẫn với một sarcoma thực thụ.
Các hội chứng di truyền liên quan đến ung thư biểu mô tế bào thận – Vai trò của xét nghiệm sinh học phân tử
Ung thư thận không phải là một bệnh đơn lẻ mà bao gồm nhiều dạng phụ khối u (tumor subtypes) dựa trên kiểu gen (genotype). Trong hơn hai thập kỷ qua, các nghiên cứu về những gia đình có ung thư biểu mô tế bào thận di truyền đã giúp xác định một số hội chứng ung thư thận di truyền cùng với các gen gây bệnh liên quan, qua đó làm sáng tỏ nhiều con đường sinh học khác nhau trong cơ chế sinh ung thư thận.
Năm 1993, gen ức chế khối u VHL (von Hippel-Lindau) liên quan đến bệnh von Hippel-Lindau di truyền và phần lớn các trường hợp ung thư biểu mô tế bào sáng (clear cell RCC, ccRCC) không di truyền đã được xác định. Sau đó, các đột biến hoạt hóa (activating mutations) được phát hiện trong proto-oncogene MET ở bệnh nhân mắc ung thư biểu mô tế bào thận nhú di truyền (hereditary papillary renal carcinoma - HPRC). Gần đây hơn, các đột biến mầm (germline mutations) trong genfumarate hydratase (FH), một enzyme thuộc chu trình Krebs, được xác định là nguyên nhân gây hội chứng leiomyomatosis di truyền và ung thư tế bào thận (HLRCC). Bên cạnh đó, đột biến gen FLCN cũng được tìm thấy ở bệnh nhân mắc hội chứng Birt–Hogg–Dubé (BHDsyndrome).
Ngoài ra, các đột biến mầm trong các gen mã hóa các tiểu đơn vị B, C và D của enzyme succinate dehydrogenase (SDHB, SDHC, SDHD) cũng được phát hiện ở những bệnh nhân mắc ung thư thận gia đình (familial renalcancer). Việc phát hiện ra các gen liên quan đến ung thư thận di truyền đã giúp phát triển các xét nghiệm di truyền chẩn đoán, hỗ trợ phát hiện sớm ở những người có nguy cơ cao và cải thiện tiên lượng bệnh.
Bảng 3. Các hội chứng ung thư thận di truyền
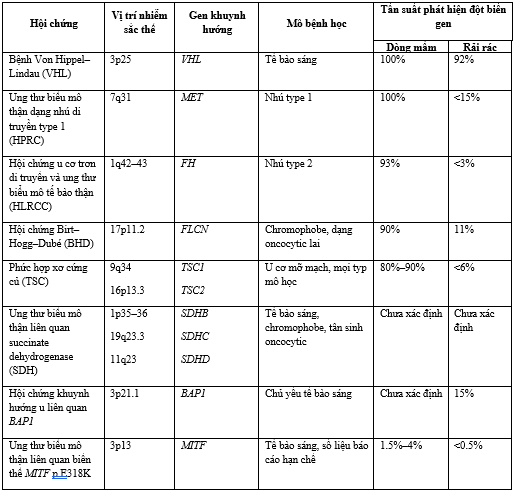
Hình 13. Tóm tắt vai trò của các đột biến gene trong cơ chế bệnh sinh gây ra ung thư biểu mô tế bào thận di truyền và không di truyền(hình tròn hồng: các gene đột biến gây ra RCC di truyền; hình tròn xanh lá: các gene đột biến gây ra RCC lẻ tẻ – không di truyền; mũi tên đầu tù màu đỏ: điều hòa âm tính/ ức chế; mũi tên đầu nhọn màu đen: điều hòa dương tính/ kích thích;xanh lam: các protein chức năng).
Cơ sở di truyền của ung thư thận. Mười ba gen gây khuynh hướng ung thư thận, bao gồm VHL, MET, FLCN, BAP1, TFE3, TFEB, MITF, TSC1, TSC2, FH, SDHB, SDHC và SDHD, đã được xác định chủ yếu thông qua các nghiên cứu về các hội chứng ung thư thận di truyền. Các gen này tương tác với nhau thông qua các con đường cảm nhận oxy, dinh dưỡng và năng lượng chung; khi các con đường này bị rối loạn điều hòa, chúng làm tăng nguy cơ phát triển ung thư thận. Việc hiểu biết ngày càng sâu sắc về các cơ chế phân tử mà qua đó các gen này tương tác trong các con đường nói trên đang tạo điều kiện cho sự phát triển các thuốc điều trị nhắm trúng đích, mang lại lợi ích cho bệnh nhân ung thư thận.
Ung Thư Biểu Mô Tế Bào Thận Tế Bào Sáng (ccRCC)
Bệnh Von Hippel-Lindau và Ung Thư Biểu Mô Tế Bào Thận Tế Bào Sáng
Ung thư biểu mô tế bào sáng của thận (Clear cell renal cell carcinoma - ccRCC) có thể xảy ra dưới dạng gia đình, di truyền hoặc lẻ tẻ (không di truyền). Dạng di truyền được nghiên cứu nhiều nhất của ccRCC là bệnh von Hippel-Lindau (VHL). Đây là một rối loạn tân sinh đa hệ thống di truyền trội trên nhiễm sắc thể thường (autosomal-dominant inherited multisystem neoplastic disorder), đặc trưng bởi các khối u tế bào sáng ở thận, u mạch võng mạc, u mạch máu hệ thần kinh trung ương, u tủy thượng thận (pheochromocytoma), u túi nội bạch huyết, u tế bào đảo tụy và các nang ở tụy và thận. Bệnh VHL có tỷ lệ mắc khoảng 1/36.000 người và thường khởi phát trong độ tuổi từ 20 đến 40, với gần như hoàn toàn biểu hiện bệnh ở tuổi 70. Khoảng 30% bệnh nhân VHL phát triển khối u thận hai bên, đa ổ với mô bệnh học tế bào sáng, có nguy cơ di căn khi đạt kích thước 3 cm. Belzutifan, một chất ức chế HIF 2-α, gần đây đã được FDA phê duyệt để điều trị bệnh nhân ccRCC liên quan đến VHL, những người đang được cân nhắc phẫu thuật cắt bỏ. Trong một nghiên cứu quan trọng, tỷ lệ đáp ứng khách quan đạt 50% đối với tổn thương thận, và đáng chú ý là chỉ có một bệnh nhân cần can thiệp giảm kích thước thận so với 250 ca phẫu thuật trước khi liệu pháp này được áp dụng. Nghiên cứu này đặt nền tảng cho việc sử dụng liệu pháp toàn thân trong quản lý ung thư tế bào thận khu trú.
Di truyền học của hội chứng Von Hippel-Lindau (VHL):
Việc nhân bản địnhvị (positional cloning) trên các gia đình có bệnh VHL đã xác định vị trí của gene VHL trên nhiễm sắc thể 3p25-26, dẫn đến việc phát hiện gene VHL vào năm 1993. VHL là một gene ức chế khối u điển hình theo mô hình "two-hit",trong đó cả hai bản sao của gene VHL phải bị bất hoạt để khối u khởi phát. Cơ chế bất hoạt này thường xảy ra thông qua mất tính dị hợp tử (Loss ofheterozygosity - LOH), đột biến soma hoặc methyl hóa alen còn lại. Các đột biến mầm (germline mutations) trong gene VHL có thể thuộc toàn bộ phổ đột biến và được tìm thấy trên toàn bộ gene. Dựa trên nguy cơ phát triển pheochromocytoma cũng như mức độ nguy cơ mắc ung thư biểu mô tế bào thận (RCC) cao hay thấp, các phân nhóm VHL đã được thiết lập với mối liên hệ rõ ràng giữa kiểu gen - kiểu hình (genotype-phenotype associations).
Chuyển đoạn nhiễm sắc thể 3p dòng mầm và ung thư biểu mô tế bào thận dạng tế bào sáng
Một dạng khác của ung thư biểu mô tế bào sáng di truyền (hereditary clear cell RCC) được xác định lần đầu tiên vào năm 1979 bởi Cohen et al., khi mô tả một gia đình có chuyển đoạn cân bằng t(3;8)(p14;q24) mang tính chất di truyền, liên quan đến sự xuất hiện của các khối u thận tế bào sáng hai bên, đa ổ. Trong các khối u từ gia đình này,người ta phát hiện sự mất nhiễm sắc thể dẫn xuất mang đoạn 3p cùng với các đột biến soma khác nhau trên bản sao còn lại của gene VHL, dẫn đến mô hình sinh uba bước (three-step tumorigenesis model) được đề xuất: (1) di truyền chuyển đoạn nhiễm sắc thể bẩm sinh (constitutional translocation), (2) mất nhiễm sắc thể dẫn xuất mang đoạn 3p25, và (3) đột biến trên bản sao VHL hoang dã (wild-type VHL)còn lại, dẫn đến bất hoạt cả hai bản sao của gene VHL và làm tăng nguy cơ phát triển ung thư biểu mô tế bào sáng của thận (clear cell RCC).
Hội Chứng BAP1 Liên Quan Đến Nguy Cơ Ung Thư: Ung Thư Biểu Mô Tế Bào Thận Dạng Tế Bào Sáng
Đột biến BAP1 đã được phát hiện trong dòng mầm (germline) của một số ít gia đình có yếu tố di truyền làm tăng nguy cơ mắc ung thư biểu mô tế bào thận dạng tế bào sáng(ccRCC). Ngoài ra, những người mang đột biến dị hợp tử (heterozygous mutation)của BAP1 cũng có nguy cơ cao phát triển các loại khối u khác, bao gồm u hắc tố màng bồ đào (uveal melanoma), u hắc tố da (cutaneous melanoma) và u trung biểu mô ác tính (malignant mesothelioma). Trong các khối u từ bệnh nhân mang đột biến BAP1, người ta đã phát hiện hiện tượng mất dị hợp tử (LOH - Loss of Heterozygosity), khẳng định rằng BAP1 là một gen ức chế khối u điển hình theo mô hình hai cú đánh (two-hit tumor suppressorgene). Những bệnh nhân mang đột biến BAP1 thường có khởi phát ung thư sớm, đi kèm với một dạng RCC có tính xâm lấn cao, liên quan đến tiên lượng xấu và tỷ lệ sống thấp. Hiện nay, chưa có thử nghiệm lâm sàng nào được thiết kế riêng để đánh giá các liệu pháp tiềm năng cho RCC liên quan đến BAP1. Tuy nhiên, trong nghiên cứu COMPARZ, ở nhóm bệnh nhân RCC tiến triển hoặc di căn chưa điều trị trước đó, những người có đột biến BAP1 có thời gian sống không tiến triển bệnh (PFS) và thời gian sống toàn bộ (OS) thấp hơn khi điều trị bằng liệu pháp nhắm trúng đích VEGF.
Ung thư biểu mô tế bào thận dạng tế bào sáng (ccRCC) không di truyền: Gen VHL
Đột biến soma của gen VHL kèm theo mất alen kiểu dại (wild-type allele) được tìm thấy trong lên đến 92% các khối u ở bệnh nhân ung thư biểu mô tế bào thận dạng tế bào sáng (ccRCC- clear cell renal cell carcinoma). Tuy nhiên, đột biến VHL không được phát hiện trong các loại ung thư thận khác như ung thư thận thể nhú (papillary RCC), thể kỵ sắc (chromophobe RCC), ống góp (collecting duct RCC), tủy thận (medullary RCC)và các dạng khác.
Chức năng của protein VHL (pVHL)
Protein VHL đóng vai trò quan trọng trong điều hòa yếu tố cảm ứng thiếu oxy (HIF - hypoxia-inducible factor). pVHL liên kết với Elongin C, Elongin B và Cullin 2, tạo thành phức hợp E3 ubiquitin ligase, giúp đánh dấu HIF để phân hủy thông qua hệ thống ubiquitin-proteasome theo cơ chế nhạy cảm với nồng độ oxy. Trong điều kiện bình thường (normoxia), HIF-α bị hydroxyl hóa tại các gốc proline quan trọng bởi prolyl hydroxylase (PHD). Sau đó, pVHL liên kết với HIF-α đã bị hydroxyl hóa, đánh dấu HIF-α để bị ubiquitin hóa và phân hủy bởi phức hợp E3 ligase. Ngược lại, trong điều kiện thiếu oxy (hypoxia) hoặc khi pVHL bị đột biến, làm mất khả năng liên kết với HIF-α hoặc Elongin C, HIF-α tích lũy và kích hoạt phiên mã nhiều gen quan trọng trong quá trình sinh mạch máu (EPO, VEGF), tăng sinh tế bào (PDGFβ, TGFα) và chuyển hóa glucose yếm khí (GLUT-1). Đặc biệt, HIF-2α (thay vì HIF-1α) được cho là yếu tố then chốt trong sinh bệnh học của ung thư biểu mô tế bào thận dạng tế bào sáng.
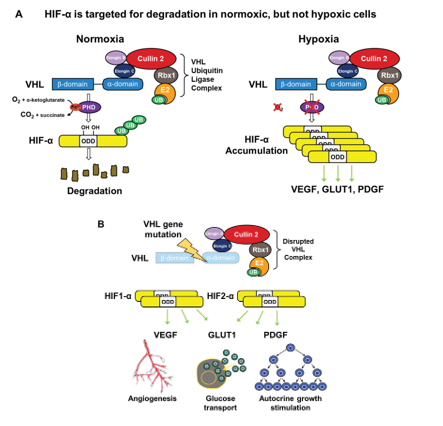
Hình 14. Chức năng sinh lý của protein VHL (hình A) và đột biến gene gây mất protein VHL trong cơ chế bệnh sinh của ung thư biểu mô tế bào thận tế bào sáng (hình B)
Phức hợp ubiquitin ligase E3 của von Hippel–Lindau (VHL) có chức năng nhắm đích yếu tố cảm ứng thiếu oxy alpha (hypoxia-inducible factor-α, HIF-α) để thoái hóa thông qua cơ chế gắn ubiquitin. A: Trong điều kiện oxy bình thường, HIF-α bị hydroxyl hóa tại các vị trí proline then chốt bởi enzyme HIF prolyl hydroxylase (PHD), quá trình này cần oxy phân tử, α-ketoglutarate và sắt làm các đồng cơ chất. Sau đó, protein VHL(pVHL) có thể nhận diện và gắn vào HIF-α đã được hydroxyl hóa, cho phép phức hợpE3 ubiquitin ligase của VHL thực hiện ubiquitin hóa và dẫn tới sự thoái hóa của HIF-α tại proteasome. Trong điều kiện thiếu oxy, PHD không thể hoạt động hiệu quả, pVHL không nhận diện được HIF-α, dẫn đến sự tích tụ của HIF-α và làm tăng biểu hiện các gen đích của HIF như VEGF, GLUT1 và PDGF, những yếu tố hỗ trợ sự phát triển khối u và tân sinh mạch. B: Khi gen VHL bị đột biến và pVHL không còn khả năng gắn với HIF-α, sự ổn định của HIF-α dẫn đến tăng điều hòa phiên mã các gen đích của HIF
Các gen đột biến bổ sung trong ung thư biểu mô tế bào thận dạng tế bào sáng
Các nghiên cứu hệ gen quy mô lớn nhằm xác định cơ sở di truyền của ung thư biểu mô tế bào thận dạng tế bào sáng đã phát hiện ra các biến đổi di truyền trong các gen quan trọng đối với việc duy trì trạng thái nhiễm sắc thể. Các gen đột biến đáng kể được xác định trong ung thư biểu mô tế bào thận dạng tế bào sáng thể lẻ tẻ, ngoài VHL, bao gồm PBRM1, mã hóa một tiểu đơn vị của phức hợp tái cấu trúc nhiễm sắc thể PBAF SWI/SNF, gen methyl transferase histone SETD2, gen khử methyl histone KDM5C, vàgen ức chế khối u mới BAP1, mã hóa một enzyme deubiquitinase histone.
Ung thư biểu mô tế bào thận dạng nhú (Papillary Renal Cell Carcinoma - PRCC)
Ung thư biểu mô tế bào thận dạng nhú di truyền: Type 1
Tương tự như ung thư biểu mô tế bào thận thể sáng (clear cell RCC), ung thư biểu mô tế bào thận dạng nhú (papillary RCC) có thể xảy ra ở cả dạng di truyền và dạng lẻ tẻ. Ung thư biểu mô tế bào thận dạng nhú di truyền (Hereditary Papillary Renal Carcinoma - HPRC) là một hội chứng ung thư di truyền trội trên nhiễm sắc thể thường, trong đó các cá nhân mắc bệnh có nguy cơ phát triển nhiều khối u dạng nhú loại 1 ở cả hai thận. HPRC thường xuất hiện ở độ tuổi 60-70, mặc dù các trường hợp khởi phát sớm đã được ghi nhận, và có tỷ lệ thâm nhiễm gần như hoàn toàn ở tuổi 80. Đây là một rối loạn hiếm gặp, với chưa đến 35 gia đình mắc bệnh được báo cáo trên toàn thế giới.
Ung thư biểu mô tế bào thận dạng nhú di truyền: Gen tiền ung thư MET
Năm 1994, Zbar etal. đã mô tả một gia đình ba thế hệ có các khối u thận dạng nhú đa ổ, hai bên,được di truyền theo kiểu trội trên nhiễm sắc thể thường (autosomal-dominant).75 Rối loạn này không liên quan đến mất đoạn nhiễm sắc thể 3p, gợi ý sự tồn tại của một gen tiền đề mới gây ung thư thận và đại diện cho dạng di truyền của ung thư biểu mô tế bào thận dạng nhú (papillary renal cell carcinoma - PRCC) lẻ tẻ. Schmidt et al. đã xác định vị trí gen gây bệnh HPRC trên nhiễm sắc thể 7q31.1-34 thông qua phân tích liên kết di truyền và phát hiện các đột biến sai nghĩa (missense mutations) tại miền tyrosine kinase của gen tiền ung thư MET (MET proto-oncogene) ở các thành viên gia đình bị ảnh hưởng.40
Hệ quả chức năng của các đột biến MET
Gen tiền ung thư MET mã hóa thụ thể tyrosine kinase của yếu tố tăng trưởng tế bào gan/yếu tố phân tán (HGF/SF). Khi HGF liên kết với MET, quá trình tự phosphoryl hóa xảy ra tại các tyrosine quan trọng trong vùng tyrosine kinase nội bào, dẫn đến sự tuyển mộ nhiều phân tử trung gian tham gia vào các con đường tín hiệu xuôi dòng. Các con đường này điều hòa nhiều quá trình tế bào quan trọng như tăng trưởng, hình thái phân nhánh, biệt hóa và tăng sinh xâm lấn.
Tất cả các đột biến MET được xác định trong HPRC đều thuộc dạng đột biến sai nghĩa (missense) và được dự đoán có khả năng hoạt hóa liên tục MET kinase mà không cần kích thích bởi ligand. Đặc điểm điển hình của khối u thận dạng nhú là trisomy nhiễm sắc thể 7, trong đó có sự nhân đôi không ngẫu nhiên của nhiễm sắc thể 7 chứa alen đột biến MET. Hiện tượng này đã được ghi nhận trong các khối u thận nhú từ bệnh nhân HPRC. Việc có hai bản sao MET đột biến có thể mang lại lợi thế tăng sinh, góp phần vào sự tiến triển của khối u.
Ung thư biểu mô tế bào thận thể nhú loại 1 không di truyền (Sporadic Type 1 pRCC)
Các nghiên cứu ban đầu về ung thư biểu mô tế bào thận thể nhú (PRCC) chưa phân loại lẻ tẻ đã báo cáo rằng đột biến trình tự mã hóa MET xuất hiện trong khoảng 13% số ca bệnh, cho thấy vai trò tương đối nhỏ của đột biến MET trong các khối u PRCC lẻ tẻ không di truyền. Tuy nhiên, nghiên cứu trên 161 khối u PRCC lẻ tẻ của The Cancer Genome Atlas Research Network đã phát hiện rằng 81% số ca PRCC loại 1 có bất thường liên quan đến MET, bao gồm đột biến trình tự (18,6%), biến thể cắt nối thay thế (5,3%), hợp nhất gen MET (4%), và khuếch đại MET do tăng số bản sao nhiễm sắc thể 7. Những phát hiện này nhấn mạnh vai trò quan trọng của MET trong cơ chế bệnh sinh của PRCC loại 1 lẻ tẻ.
Hỗ trợ cho vai trò nổi bật của MET trong PRCC loại 1, một nghiên cứu lâm sàng pha II đã đánh giá hiệu quả của foretinib (trước đây gọi là XL880)—một chất ức chế đa kinase nhắm vào MET cùng các tyrosine kinase khác—trên bệnh nhân mắc PRCC lẻ tẻ dạng di căn, đa ổ hoặc hai bên, cũng như bệnh nhân HPRC mang đột biến mầm MET. Kết quả cho thấy foretinib có hiệu quả trong điều trị PRCC tiến triển với tỷ lệ đáp ứng khách quan (ORR) đạt 13,5% và thời gian sống không tiến triển (PFS) trung bình là 9,3 tháng, trong đó nhóm bệnh nhân có đột biến mầm MET có đáp ứng cao nhất.
Ung thư biểu mô tế bào thận liên quan đến họ gen Microphthalmia (MiT): TFE3, TFEB, và MITF
Ung thư biểu mô tế bào thận do chuyển đoạn Xp11.2 thường có cấu trúc nhú với bào tương sáng hoặc ưa eosin. Đây là một dạng ung thư hiếm gặp ở người lớn (<5%) nhưng chiếm khoảng 40% các trường hợp ung thư thận ở trẻ em. Các chuyển đoạn trong ung thư biểu mô tế bào thận dạng nhú xảy ra ở vùng Xp11.2 và 1q21.2 tạo ra một tổ hợp gen dung hợp giữa PRCC và TFE3—một yếu tố phiên mã thuộc họ microphthalmia (MiT).Protein dung hợp PRCC-TFE3 có hoạt tính phiên mã mạnh hơn so với TFE3 bình thường, có thể thúc đẩy sự phát triển của ung thư biểu mô tế bào thận dạng nhú.
Ngoài PRCC-TFE3,ít nhất năm tổ hợp gen dung hợp khác liên quan đến TFE3 đã được mô tả, dẫn đến sự rối loạn hoạt động phiên mã của TFE3. Một thành viên khác trong họ MiT, TFEB, cũng có thể bị dung hợp với Alpha gene trong các khối u thận mang chuyển đoạn nhiễm sắc thể t(6;11)(p21;q13).
Gần đây, ung thư thận liên quan đến thành viên thứ ba của họ MiT, MITF, cũng đã được ghi nhận. Đột biến sai nghĩa dòng mầm MITF (c.952G→A; p.E318K) được phát hiện với tần suất cao hơn ở những thành viên trong gia đình mắc melanoma, ung thư thận, hoặc cả hai, làm tăng nguy cơ mắc các bệnh này lên gấp năm lần. Codon 318 của MITF nằm trong vị trí gắn kết với small ubiquitin-like modifier (SUMO), và đột biến này làm suy giảm nghiêm trọng quá trình SUMOylation (ức chế) của MITF. Hậu quả là MITF có hoạt tính phiên mã mạnh hơn đối với HIF-α và các mục tiêu khác, thúc đẩy quá trình sinh ung thư.
Các khối u từ bệnh nhân mang đột biến MITF p.E318K cho thấy MITF tập trung trong nhân, nơi nó phối hợp với các thành viên khác của họ MiT để điều hòa sự biểu hiện của các gen mục tiêu liên quan đến tự thực bào (autophagy), sinh tổng hợp lysosome, sự sống còn của tế bào và bảo vệ chống lại stress oxy hóa.
Bệnh u cơ trơn di truyền và ung thư biểu mô tế bào thận
Bệnh u cơ trơn di truyền và ung thư biểu mô tế bào thận (HLRCC) là một hội chứng ung thư di truyền theo kiểu trội trên nhiễm sắc thể thường. Những người mắc bệnh này có nguy cơ phát triển u cơ trơn da, u cơ trơn tử cung, và một dạng ung thư biểu mô tế bào thận thể nhú type 2 có tính chất xâm lấn cao. Các khối u thận trong HLRCC thường đơn độc và khu trú một bên thận, có thể xuất hiện sớm với tỷ lệ 14-18% ở những người bị ảnh hưởng. Những khối u này có thể phát triển theo hướng xâm lấn mạnh, di căn sớm,và gây tử vong trong vòng 5 năm sau chẩn đoán.
HLRCC: Gen Fumarate Hydratase
Phân tích liên kết di truyền đã xác định vị trí của locus bệnh đối với bệnh đa u cơ trơn da và tử cung (multiple cutaneous and uterine leiomyomata – MCUL) tại nhiễm sắc thể 1q42-43. Tuy nhiên, mối liên quan với ung thư thận chưa được ghi nhận cho đến khi phát hiện sự liên kết với nhiễm sắc thể 1q ở hai gia đình người Phần Lan mắc MCUL, trong đó các thành viên có khối u thận thể nhú loại 2 đơn độc với đặc điểm rất xâm lấn. Do đó, bệnh này được đổi tên thành hội chứng u cơ trơn di truyền và ung thư tế bào thận (hereditary leiomyomatosis and renal cell carcinoma –HLRCC). Sau đó, các đột biến dòng mầm được phát hiện ở các thành viên gia đình mắc HLRCC trong gen fumarate hydratase (FH), mã hóa một enzyme trong chu trình Krebs có chức năng chuyển fumarate thành malate. Các đột biến FH trong HLRCC bao gồm đột biến sai nghĩa (missense mutations), chủ yếu ảnh hưởng đến các codon bảo tồn theo tiến hóa, cũng như đột biến dịch khung (frameshiftmutations), đột biến vô nghĩa (nonsense mutations), và đột biến vị trí ghép nối (splice-site mutations), dẫn đến sự cắt ngắn protein FH một cách bất thường.Ngoài ra, còn có các mất đoạn một phần hoặc toàn bộ gen FH. Các đột biến được tìm thấy trên toàn bộ chiều dài của gen FH, và chưa có mối liên quan rõ ràng giữa kiểu gen và kiểu hình (genotype–phenotype associations). Gen FH hoạt động như một gen ức chế khối u cổ điển (tumor suppressor gene), với hiện tượng mất hoặc đột biến soma của alen FH bình thường xuất hiện với tần suất cao trong các khối u thận, cũng như trong u cơ trơn da và tử cung. Tuy nhiên, các đột biến FH hiếm khi được phát hiện trong u cơ trơn tử cung và da dạng lẻ tẻ, hoặc trong ung thư tế bào thận không liên quan đến HLRCC.
Hậu quả chức năng của đột biến gen FH
Các đột biến FH gây ra sự suy giảm nghiêm trọng hoạt động của enzyme FH trong các dòng tế bào lymphoblastoid từ bệnh nhân HLRCC. Các đột biến sai nghĩa (missense mutations) liên quan đến HLRCC làm giảm hoạt động FH đáng kể hơn so với các đột biến gây mất đoạn (truncating mutations). Điều này gợi ý rằng các monomer FH đột biến có thể hoạt động theo cơ chế áp chế trội âm (dominant negative effect), làm thay đổi cấu trúc bình thường của tetramer FH. Mất hoạt động FH trong HLRCC dẫn đến tích lũy fumarate, chất này có thể ức chế cạnh tranh enzym HIF prolyl hydroxylase, làm ổn định HIF-1α. Sự tăng cường biểu hiện các gen cảm ứng bởi HIF góp phần vào tính chất xâm lấn mạnh của các khối u thận liên quan đến HLRCC.
Một hậu quả khác của sự tích lũy fumarate trong khối u HLRCC là kích hoạt con đường tín hiệu chống oxy hóa do NRF2 (nuclear factor erythroid-derived 2–like 2). Bình thường,protein KEAP1 (Kelch-like ECH-associated protein 1) hoạt động như một cảm biến electrophile, nhận diện và liên kết với NRF2, tạo điều kiện để NRF2 bị phân hủy bởi hệ thống E3 ubiquitin ligase dựa trên CUL3 (cullin-3).
Tuy nhiên, fumarate tích lũy có thể hoạt động như một electrophile, phản ứng với các nhóm cysteine trên KEAP1 (một quá trình gọi là succination), dẫn đến thay đổi cấu trúc KEAP1 và ức chế liên kết giữa KEAP1 và NRF2. Hậu quả là NRF2 không bị phân hủy, trở nên hoạt hóa phiên mã, điều chỉnh sự biểu hiện của các gen đích thông qua yếu tố đáp ứng chống oxy hóa (ARE – antioxidant response elements) trong vùng promoter của chúng.
Ung thư biểu mô tế bào thận thể nhú loại 2 (Type 2 PRCC) không di truyền
Nghiên cứu của The Cancer Genome Atlas Research Network đã xác nhận rằng ung thư biểu mô tế bào thận thể nhú loại 1 (Type 1 PRCC) và loại 2 (Type 2 PRCC) có sự khác biệt rõ rệt về mặt lâm sàng và sinh học. Trong khi Type 1 PRCC liên quan đến biến đổi gen MET, dẫn đến hoạt hóa con đường tín hiệu MET và thường có diễn tiến bệnh chậm hơn, thì Type 2 PRCC lại đặc trưng bởi sự bất hoạt CDKN2A (25%), đột biến SETD2, tái tổ hợp gen TFE3/TFEB, và tăng biểu hiện (hoạt hóa) con đường tín hiệu NRF2-antioxidant response element (ARE). Ngoài ra, một phân nhóm của Type 2PRCC còn có kiểu hình methyl hóa CpG island (CIMP), liên quan đến methyl hóa quá mức hoặc mất promoter của CDKN2A, đột biến FH, và một kiểu hình khối u có mức độ xâm lấn cao nhất với tiên lượng sống kém nhất.
Ung thư biểu mô tế bào thận thể sắc bào (Chromophobe Renal Cell Carcinoma - ChRCC)
Hội chứng Birt-Hogg-Dubé (Birt-Hogg-Dubé Syndrome - BHD)
Hội chứng Birt-Hogg-Dubé (BHD) là một hội chứng ung thư di truyền hiếm gặp theo kiểu trội trên nhiễm sắc thể thường, đặc trưng bởi sự xuất hiện của các u lành tính của nang lông (fibrofolliculoma), các nang phổi (pulmonary cysts), tràn khí màng phổi tự phát, và nguy cơ mắc ung thư thận cao gấp bảy lần so với dân số chung. Các biểu hiện phổ biến nhất của BHD là fibrofolliculomas (>85%) và nang phổi(>70%). Bệnh nhân BHD có thể phát triển các khối u thận với kiểu hình mô bệnh học khác nhau, phổ biến nhất là ung thư biểu mô tế bào thận thể sắc bào (chromophobe renal carcinoma) và u lai dạng tế bào ưa acid (hybrid oncocytictumors), với tỷ lệ mắc từ 12% đến 34% (tuổi trung bình khởi phát từ 48 đến 50 tuổi). Mặc dù hiếm gặp, nhưng một số trường hợp ung thư thận trong BHD có thể tiến triển di căn.
Hội chứng Birt-Hogg-Dubé: Gen FLCN
Phân tích liên kết di truyền trên các gia đình mắc BHD đã xác định vùng bệnh lý nằm trên nhánh ngắn của nhiễm sắc thể 17 và sau đó phát hiện các đột biến trên một gen mới là FLCN. Các đột biến thường gặp trong FLCN dẫn đến sự suy giảm chức năng protein folliculin, bao gồm đột biến chèn/xóa, đột biến vô nghĩa, đột biến vị trí ghép nối (splice-site mutations), cũng như một số ít trường hợp đột biến sai nghĩa và mất đoạn một phần của gen. Các đột biến phân bố trên toàn bộ chiều dài của gen FLCN mà không có sự tương quan rõ ràng giữa kiểu gen và kiểu hình. Nghiên cứu của Vocke et al. đã phát hiện các đột biến soma dạng "hit thứ hai" hoặc mất dị hợp tử (LOH - loss of heterozygosity) trong 70% các khối u thận từ bệnh nhân BHD, cho thấy vai trò của FLCN như một gen ức chế khối u. Các đột biến FLCN hiếm khi xuất hiện trong ung thư biểu mô tế bào thận thể sắc bào (11%) và rất ít gặp trong các biến thể mô bệnh học khác của ung thư thận.
Chức năng của Protein Birt-Hogg-Dubé
Gen FLCN mã hóa protein folliculin (FLCN), một protein chưa được xác định rõ vai trò chức năng.Nghiên cứu về chức năng của FLCN đã phát hiện hai protein tương tác mới là FNIP1 và FNIP2, có liên kết với adenosine monophosphate-activated proteinkinase (AMPK)—một cảm biến năng lượng và chất ức chế con đường mTOR (một yếu tố điều hòa chính của quá trình dịch mã protein và tăng sinh tế bào). Do đó, FLCN có thể đóng vai trò điều hòa con đường tín hiệu PI3K-AKT-mTOR. Tuy nhiên, dữ liệu từ các mô hình động vật bị mất FLCN và từ các khối u thận của bệnh nhân BHD lại cho thấy kết quả mâu thuẫn giữa kích hoạt và ức chế mTOR, dẫn đến giả thuyết rằng FLCN có thể điều hòa mTOR theo bối cảnh cụ thể.Gần đây, FNIP1 và FNIP2 cũng được phát hiện có vai trò như đồng chaperone của protein HSP90, giúp ổn định protein FLCN. Cấu trúc tinh thể của FLCN đã cho thấy sự tương đồng với protein DENN1B, cho thấy FLCN có thể chứa miền DENN ở C-terminal và miền Longin ở N-terminal, những đặc điểm của một yếu tố trao đổi nucleotide guanine (GEF) cho RabGTPases. Dù vai trò GEF của FLCN chưa được nghiên cứu sâu, nhưng phức hợp FLCN/FNIP1/FNIP2 đã được chứng minh hoạt động như một protein kích hoạt GTPase (GAP) đối với RagC/D trên bề mặt lysosome. Khi có sẵn axit amin, dị thể hoạt động RagA^GTP hoặc RagB^GTP kết hợp với RagC^GDP hoặc RagD^GDP sẽ tuyển dụng phức hợp mTORC1 đến bề mặt lysosome để được kích hoạt bởi Rheb. Ngoài ra, FLCN còn điều hòa hoạt động của các yếu tố phiên mã TFE3 và TFEB, những yếu tố chính kiểm soát sự sinh tổng hợp lysosome. FLCN có thể giữ TFE3 trong bào tương thông qua cơ chế phosphoryl hóa qua trung gian mTORC1. Khi FLCN bị bất hoạt, TFEB di chuyển vào nhân và kích hoạt phiên mã các gen đích, bao gồm cả RagC và RagD, tạo thành một vòng lặp tăng cường hoạt động của mTORC1. Việc bất hoạt TFEB đã được chứng minh có thể cứu vãn kiểu hình tổn thương thận trong mô hình chuột Flcnknockout, gợi ý rằng TFEB có thể là một trong những yếu tố thúc đẩy chính của ung thư thận liên quan đến BHD. Cuối cùng, FLCN còn tham gia vào nhiều quá trình khác như điều hòa tín hiệu TGF-β, điều hòa HIF-α và các gen đích của nó,chức năng của trung thể (ciliogenesis), điều hòa PGC-1α và sinh tổng hợp ty thể, kết dính tế bào, và điều hòa trạng thái tế bào gốc phôi.
Ung thư biểu mô tế bào thận thể sắc bào không di truyền (Sporadic Chromophobe RCC)
Đặc điểm phân tử của ung thư biểu mô tế bào thận thể sắc bào không di truyền (sporadic chromophobe RCC) do The Cancer Genome Atlas Research Network phân tích đã cho thấy mất một bản sao của toàn bộ hoặc hầu hết các nhiễm sắc thể 1, 2, 6, 10, 13 và 17 trong phần lớn các trường hợp (86%). Phân tích không xác định được đột biến FLCN, nhưng phát hiện rằng TP53 (32%) và PTEN (9%) là hai gen bị đột biến có ý nghĩa trong chromophobe RCC. Các tái sắp xếp bộ gen được tìm thấy ở 12% trường hợp, dẫn đến điểm gãy cấu trúc trong vùng khởi động (promoter) của TERT, có liên quan đến sự gia tăng mạnh mẽ mức độ biểu hiện của TERT. Ngoài ra, phân tích biểu hiện gen và mtDNA cho thấy sự gia tăng số bản sao của bộ gen ty thể và biểu hiện của PPARGC1A, một chất điều hòa chức năng ty thể, gợi ý rằng có sự gia tăng sinh tổng hợp ty thể trong ung thư biểu mô tế bào thận thể sắc bào không di truyền.
Các loại ung thư biểu mô tế bào thận (RCC) khác
Phức hợp xơ cứng củ
Phức hợp xơ cứng củ (Tuberous Sclerosis Complex - TSC) là một rối loạn di truyền trội trên nhiễm sắc thể thường, ảnh hưởng đến nhiều hệ cơ quan ở cả trẻ em và người lớn. Bệnh đặc trưng bởi sự xuất hiện của u sợi mạch máu ở mặt (facial angiofibromas), u mỡ cơ mạch thận (renal angiomyolipomas - AML), bệnh bạch mạch cơ mạch phổi (lymphangiomyomatosis) và các biểu hiện thần kinh nghiêm trọng. Tuy nhiên, TSC có biểu hiện lâm sàng rất đa dạng, và nhiều bệnh nhân chỉ có triệu chứng tối thiểu. Biểu hiện tại thận thường gặp nhất của TSC là AML đa ổ hai bên, đây là các khối u lành tính gồm mạch máu bất thường, tế bào cơ trơn chưa trưởng thành và tế bào mỡ. Nguy cơ suốt đời mắc ung thư thận ở bệnh nhân TSC dao động từ 2%đến 4%, tương đương với dân số chung. Loại ung thư biểu mô tế bào thận (RCC) phổ biến nhất trong TSC là thể tế bào sáng (clear cell RCC), tuy nhiên cũng có một số báo cáo hiếm gặp về RCC thể nhú (papillary RCC), RCC thể sắc tố (chromophobe RCC) và u tế bào ưa acid (oncocytoma). TSC được gây ra bởi đột biến ở một trong hai gen: TSC1 (mã hóa protein hamartin) hoặc TSC2 (mã hóa protein tuberin). Các đột biến này dẫn đến mất điều hòa ức chế con đường tín hiệu mTOR (mammaliantarget of rapamycin), góp phần vào sự phát triển của các khối u trong bệnh TSC.
Ung thư thận liên quan đến Succinate Dehydrogenase (SDH-Associated Renal Cancer)
Bệnh nhân có khối u thận đa ổ hai bên khởi phát sớm (<40 tuổi), có hoặc không kèm theo u hạch thần kinh đầu cổ di truyền (hereditary head and neck paragangliomas - HPGL) vàu tủy thượng thận hoặc ngoài thượng thận (pheochromocytomas), đã được ghi nhận có đột biến dòng mầm (germline mutation) ở gen SDHB. SDHB mã hóa tiểu đơn vị B của enzyme succinate dehydrogenase, một thành phần của chu trình Krebs.
Bên cạnh đó, đột biến dòng mầm ở gen SDHD, ban đầu được phát hiện có liên quan đến HPGL và sau đó là pheochromocytomas di truyền và lẻ tẻ, cũng đã được ghi nhận liên quan đến bệnh cảnh ung thư biểu mô tế bào thận (RCC) di truyền. Gần đây, hai trường hợp RCC liên quan đến SDH đã được báo cáo ở bệnh nhân mang đột biến dòng mầm SDH subunit C (SDHC).Hình thái phổ biến nhất của RCC liên quan đến SDH là khối u có đặc điểm tăng sinh bào tương ưa eosin (oncocytic neoplastic features), tuy nhiên, nhiều dạng mô bệnh học khác nhau cũng đã được mô tả. Phổ đột biến SDHB/SDHC/SDHD trong RCC liên quan đến SDH bao gồm đột biến sai nghĩa (missense), dịch khung (frameshift) và vô nghĩa (nonsense mutations).
Cơ chế bệnh sinh:
- Mất chức năng của gen SDH dẫn đến giảm hoạt động enzyme SDH, gây tích lũy succinate trong khối u thận.
- Succinate ức chế cạnh tranh cofactor α-ketoglutarate của prolyl hydroxylase (PHD), làm ức chế hoạt động của PHD.
- Khi PHD bị ức chế, HIF-α không bị thoái hóa, tích lũy trong tế bào và hoạt hóa quá trình phiên mã các gen đích của HIF-α, thúc đẩy tân sinh mạch, tăng sinh và xâm lấn khối u.
Cơ chế này tương tự như bệnh ung thư thận liên quan đến fumarate hydratase (HLRCC - Hereditary Leiomyomatosis and Renal Cell Cancer), trong đó tích lũy fumarate ức chế PHD, dẫn đến tăng hoạt hóa HIF-α.
Chẩn đoán hình ảnh
Siêu âm
Mặc dù siêu âm rất thường được chỉ định để đánh giá hệ tiết niệu, phương pháp này không nhạy và không đặc hiệu bằng chụp cắt lớp vi tính (CT) hay cộng hưởng từ (MRI). Ngoài ra, trong nhiều trường hợp,siêu âm gặp khó khăn trong việc đánh giá chính xác giai đoạn tại chỗ của bệnh.Ung thư biểu mô tế bào thận có hình ảnh siêu âm rất đa dạng; khối u có thể dạng đặc hoặc dạng nang một phần, và có thể tăng âm, đẳng âm hoặc giảm âm so với nhu mô thận xung quanh. Giả bao u đôi khi có thể quan sát được trên siêu âm dưới dạng một viền giảm âm bao quanh khối u. Mặc dù đây là một dấu hiệu tương đối đặc hiệu,độ nhạy của dấu hiệu này không cao, khoảng 20%. Việc sử dụng kỹ thuật siêu âm hài hòa (harmonic scanning) được báo cáo là có thể làm tăng độ nhạy lên tới khoảng 85%. Siêu âm có tiêm thuốc tương phản thường cho thấy tổn thương tăng sinh mạch không đồng nhất trong thì động mạch và rửa thuốc sớm ở thì muộn.
Cắt lớp vi tính
CT được sử dụng rộng rãi cả trong chẩn đoán và phân giai đoạn ung thư biểu mô tế bào thận. Trên CT không tiêm thuốc cản quang,tổn thương thường có tỷ trọng mô mềm, dao động trong khoảng 20–70 đơn vị Hounsfield. Các khối u lớn thường có những vùng hoại tử, và khoảng 30% trường hợp có vôi hóa. Trong thì vỏ–tủy, từ 25 đến 70 giây sau tiêm thuốc cản quang, ung thư biểu mô tế bào thận biểu hiện mức độ ngấm thuốc thay đổi, thường thấp hơn so với vỏ thận bình thường. Các tổn thương nhỏ có thể ngấm thuốc tương đương nhu mô vỏ thận và do đó khó phát hiện.
Nhìn chung, các khối u nhỏ thường ngấm thuốc đồng nhất, trong khi các khối u lớn hơn có hình ảnh ngấm thuốc không đều do sự hiện diện của các vùng hoại tử. Phân nhóm tế bào sáng có thể cho thấy mức độ ngấm thuốc mạnh hơn rõ rệt.
Thì vỏ–tủy cũng là thì tối ưu để đánh giá giải phẫu mạch máu, bao gồm xâm lấn tĩnh mạch thận và các biến thể động mạch, đặc biệt khi đang cân nhắc phẫu thuật cắt thận bán phần. Sự phát triển của khối u vào trong lòng hệ tĩnh mạch, đặc biệt là tĩnh mạch thận, xảy ra trong khoảng 10% trường hợp (dao động 4–15%). Tiên lượng ở những bệnh nhân có xâm lấn tĩnh mạch chủ dưới xấu hơn rõ rệt so với trường hợp chỉ xâm lấn tĩnh mạch thận, do đó việc nhận diện chính xác trên CT có ý nghĩa quan trọng.
Thì nhu mô thận, từ 80 đến 180 giây sau tiêm thuốc, là thì nhạy nhất để phát hiện các vùng ngấm thuốc bất thường.
Thì bài xuất ít giá trị hơn, nhưng vẫn quan trọng trong việc đánh giá giải phẫu hệ thống đài bể thận, đặc biệt ở những bệnh nhân có khả năng là ứng viên cho phẫu thuật cắt thận bán phần.
Chẩn đoán hình ảnh theo dõi sau điều trị thường được thực hiện bằng CT, trong đó khuyến cáo chụp hai thì ổ bụng nhằm tối đa hóa khả năng phát hiện di căn tạng đặc. Ung thư biểu mô tế bào thận thường gây di căn tăng sinh mạch, được thấy rõ nhất ở thì động mạch khi chụp vùng bụng trên.
Cộng hưởng từ
MRI không chỉ cho hình ảnh rất tốt trong đánh giá thận và phân giai đoạn tại chỗ khối u, mà còn có thể gợi ý typ mô học dựa trên sự khác biệt tín hiệu ở chuỗi T2. Trên chuỗi T1, khối u thường có tín hiệu không đồng nhất do hoại tử, xuất huyết và các thành phần đặc. Trên chuỗi T2, hình ảnh phụ thuộc vào typ mô học: ung thư biểu mô tế bào thận tế bào sáng thường tăng tín hiệu, trong khi ung thư biểu mô tế bào thận dạng nhú thường giảm tín hiệu. Trên T1 có tiêm gadolinium, khối u thường cho thấy ngấm thuốc động mạch sớm. Giả bao u, chủ yếu chỉ gặp ở các ung thư biểu mô tế bào thận độ thấp, u tuyến thận và oncocytoma, biểu hiện dưới dạng một viền giảm tín hiệu nằm giữa khối u và nhu mô thận lành kế cận.
MRI cũng hữu ích trong đánh giá huyết khối u trong tĩnh mạch thận và tĩnh mạch chủ dưới cũng như mức độ lan lên trên của huyết khối, yếu tố quan trọng trong lập kế hoạch trước phẫu thuật. Sự hiện diện của ngấm thuốc trong huyết khối giúp phân biệt huyết khối u với huyết khối đơn thuần. Việc sử dụng các chuỗi khuếch tán đã được nghiên cứu nhằm hỗ trợ phân biệt các tổn thương thận nhỏ không xác định, có thể là tổn thương viêm hoặc ác tính; cả hai đều có hạn chế khuếch tán, tuy nhiên mức độ hạn chế thường mạnh hơn trong áp xe so với khối u.
Y học hạt nhân
Xạ hình (cho thêm cơ chế + ảnh là được)
Xạ hình thận chức năng với 99mTc-DTPA hoặc 99mTc-MAG3
Nguyên lý
Diethylenetriamine pentaacetic acid (DTPA) sau khi vào máu chỉ khoảng 3-5% gắn với protein huyết tương, phần còn lại nhanh chóng được lọc bởi cầu thận và bài xuất qua nước tiểu. DTPA không được tái hấp thu và bài tiết bởi ống thận. Dùng DTPA đánh dấu bởi 99mTc, tiêm tĩnh mạch, ghi hình động, giúp đánh giá hình ảnh và chức năng thận (chức năng lọc của cầu thận).
Mercaptoacetylglycine (MAG3) sau khi vào máu nhanh chóng được gắn với protein huyết tương với tỷ lệ gắn cao 80-90%, sau đó nhanh chóng được bài xuất qua thận: lọc ở cầu thận và tiết ở ống thận, nhưng không được tái hấp thu ở ống thận. Dùng MAG3 đánh dấu bởi Tc99m, tiêm tĩnh mạch, ghi hình động, giúp đánh giá hình ảnh và chức năng thận (dòng huyết tương hiệu dụng qua thận: effective renal plasma flow - ERPF)
Đánh giá
Sự thay đổi pha mạch (tưới máu), pha bài tiết,pha bài xuất của từng thận tùy theo vị trí, kích thước, số lượng khối u thận. Xác định được chức năng của từng thận riêng rẽ, bổ sung thông tin cho việc lên kế hoạch phẫu thuật.
Xạ hình thận hình thể với 99mTc-DSMA
Nguyên lý
DMSA (Dimercaptosuccinic acid) được đánh dấu với đồng vị phóng xạ Technetium-99m. Sau tiêm tĩnh mạch, DMSA gắn vào protein huyết tương và tập trung vào nhu mô vỏ thận, đặc biệt là các tế bào ống thận.
Đánh giá
Hình ảnh ổ khuyết xạ ở thận do khối RCC không bắt DSMA, cần phân biệt với nang thận và các khối u thận khác.
Xạ hình xương với 99mTc-MDP
Nguyên lý
Ghi hình xương bằng đồng vị phóng xạ dựa trên nguyên lý là các vùng xương bị tổn thương hay vùng xương bị phá huỷ thường đi kèm với tái tạo xương mà hệ quả là tăng hoạt động chuyển hoá và quay vòng canxi và phospho. Nếu dùng các dược chất phóng xạ có chuyển hoá tương đồng với canxi và phospho (99mTc – MDP) thì chúng sẽ tập trung tại các vùng tái tạo xương cao hơn hẳn so với tổ chức xương bình thường. Như vậy những nơi xương bị tổn thương sẽ có hoạt độ phóng xạ cao hơn so với tổ chức xương lành xung quanh dễ dàng phát hiện được trên xạ hình, SPECT xương.
Đánh giá
Ung thư biểu mô tế bào thận thường di căn tại phổi, xương. Bên cạnh vai trò của CT và MRI, xạ hình xương với 99mTc-MDP giúp đánh giá tổn thương di căn xương, chẩn đoán giai đoạn bệnh trước điều trị và theo dõi đáp ứng điều trị của bệnh. Các vị trí thường gặp di căn là xương cột sống, xương sườn, xương chậu, xương sọ,…
SPECT/CT với 99mTc-Sestamibi
Cơ sở phân tử và cơ chế
Dựa trên nguyên lý“đánh dấu” những mô có xu hướng giữ lại một chất cation thân lipid đi vào tế bào nhờ chênh lệch điện thế màng. Sau khi tiêm tĩnh mạch, sestamibi khuếch tán vào tế bào và đặc biệt có khuynh hướng tích lũy trong ty thể vì ty thể có điện thế màng âm hơn so với bào tương. Những khối u thận có mật độ ty thể cao, điển hình là oncocytoma và các u thuộc nhóm oncocytic hoặc hybrid oncocytic chromophobe tumor, thường giữ sestamibi tốt và vì vậy sẽ “sáng” trên SPECT. Ngược lại, nhiều ung thư biểu mô tế bào thận, đặc biệt clear cell renal cellcarcinoma, thường bắt thấp hoặc âm tính tương đối vì không thuộc nhóm u giàu ty thể theo kiểu oncocytic, và thêm vào đó một phần tổn thương ác tính có thể có cơ chế tống xuất chất đánh dấu qua các bơm vận chuyển, làm giảm giữ thuốc trong tế bào.
Vai trò phân biệt trong RCC
Trong bối cảnh khối u thận, vai trò nổi bật nhất của ⁹⁹ᵐTc-sestamibi SPECT/CT không phải là phân giai đoạn toàn thân mà là hỗ trợ phân loại khối u đặc tại thận, nhất là các “smallrenal mass” chưa rõ bản chất trên CT hoặc MRI. Kỹ thuật này có giá trị thực hành khi mục tiêu là nhận diện nhóm u oncocytic lành tính hoặc ít ác tính để giảm khả năng phẫu thuật không cần thiết và tăng độ tin cậy khi chọn theo dõi chủ động,đồng thời hỗ trợ quyết định có nên sinh thiết hay can thiệp. Theo các tổng quan hệ thống, hiệu năng chẩn đoán của sestamibi khá cao cho mục tiêu nhận diện oncocytoma hoặc các tổn thương oncocytic tương tự, vì vậy nó được xem là một xét nghiệm bổ sung hữu ích trong thuật toán xử trí khối u thận chọn lọc.
Đặc điểm hình ảnh
U oncocytoma hoặc uoncocytic thường biểu hiện tăng bắt sestamibi rõ rệt so với nhu mô thận nền, tạo một ổ “hot” trùng với vị trí khối u trên CT. Nhiều RCC, nhất là clear cell, thường cho hình ảnh bắt thấp hơn nền hoặc chỉ bắt nhẹ, vì vậy có thể xem là “cold” tương đối so với oncocytoma trong cùng bối cảnh. Tuy nhiên cần nhớ rằng có sự chồng lấp: một số chromophobe RCC hoặc các tổn thương có thành phần oncocytic có thể bắt sestamibi, gây dương tính giả nếu mục tiêu là loại trừ ác tính tuyệt đối; ngược lại, những khối u lành nhưng không giàu ty thể hoặc có yếu tố kỹ thuật bất lợi vẫn có thể bắt thấp, gây âm tính giả.
Ưu điểm
Cung cấp thông tin sinh học chức năng mà CT/MRI thường không trực tiếp phản ánh, tức là khuynh hướng giàu ty thể và khả năng giữ chất đánh dấu của mô u. Vì nhiều khối u thận nhỏ có hình thái chồng lấp giữa lành và ác, việc có thêm một tín hiệu chức năng “nghiêng về oncocytic” có thể giúp giảm can thiệp quá mức.
Hạn chế
Sestamibi không phải chất nhắm trúng đích đặc hiệu cho RCC, nên không phù hợp để phân giai đoạn hay đánh giá toàn thân bệnh RCC, và vẫn có vùng xám do chồng lấp bắt thuốc giữa một số subtype. Ngoài ra, kết quả phụ thuộc ngưỡng đọc, chuẩn hóa quy trình SPECT/CT, và đặc điểm của khối u như kích thước, hoại tử, hoặc các yếu tố ảnh hưởng đến tín hiệu, do đó luôn cần diễn giải cùng lâm sàng và CT/MRI.
PET/CT
Vai trò của PET/CT trong chẩn đoán, điều trị và theo dõi ung thư biểu mô tế bào thận khác nhau phụ thuộc vào các chất đánh dấu (tracer) khác nhau được sử dụng trong kỹ thuật này.
18F-FDG PET/CT
Không giống như nhiều loại ung thư khác, chụp PET/CT với FDG có giá trị hạn chế trong đánh giá ung thư biểu mô tế bào thận nguyên phát, chủ yếu do sự bài tiết sinh lý của FDG qua thận, làm giảm độ tương phản giữa tổn thương thận và nhu mô thận bình thường, đồng thời có thể che lấp hoặc làm mờ các tổn thương tại thận. Tuy nhiên, một số công bố cho thấy FDG-PET có thể được sử dụng hiệu quả trong theo dõi sau phẫu thuật và đánh giá lại giai đoạn bệnh như một phương tiện bổ trợ khi các phương pháp chẩn đoán hình ảnh thông thường không cho kết quả rõ ràng, cũng như trong một số tình huống đặc biệt như phân biệt huyết khối lành tính hoặc huyết khối trơ với huyết khối u.
PET/CT với 11C-Acetate
Cơ sở phân tử và cơ chế
¹¹C-acetate là nguyên liệu đơn giản nhất dùng để tổng hợp chất béo (lipid) và sinh năng lượng. Trong cơ thể, acetate nhanh chóng đi vào tế bào và được chuyển thành acetyl-CoA.Acetyl-CoA có thể đi theo hai “con đường” chính: đi vào chu trình tạo năng lượng của ty thể, hoặc đi vào dây chuyền tổng hợp lipid để tạo màng tế bào. Tế bào ung thư thường phải xây nhiều màng tế bào mới vì chúng tăng sinh, nên một số khối u sẽ “tiêu thụ” acetate nhiều hơn mô bình thường. PET/CT ghi nhận nơi nào tích lũy ¹¹C-acetate nhiều hơn nền xung quanh thì biểu hiện bằng vùng tăng bắt phóng xạ, từ đó gợi ý mô đang hoạt động chuyển hóa theo hướng sử dụng acetate mạnh.
Vai trò trong RCC
Trong RCC, ¹¹C-acetatePET/CT chủ yếu mang vai trò “bổ sung” khi hình ảnh học hình thái (CT/MRI) chưa đủ chắc chắn, hoặc khi ¹⁸F-FDG PET/CT có khả năng bỏ sót vì một phần RCC bắt FDG không cao. Kỹ thuật này có thể hữu ích trong một số tình huống chọn lọc như đánh giá khối u thận hoặc khối dạng nang phức tạp để tăng độ tin cậy chẩn đoán,hỗ trợ định hướng quyết định theo dõi, sinh thiết hay can thiệp. Tuy nhiên, đây không phải kỹ thuật chuẩn thường quy cho phân giai đoạn RCC ở đa số cơ sở.
Đặc điểm hình ảnh của RCC với ¹¹C-acetate PET/CT
Hình ảnh thường được mô tả là ổ RCC có thể tăng bắt acetate so với nhu mô thận hoặc mô nền lân cận,thể hiện như một vùng “hot” trên PET tương ứng tổn thương trên CT. Mức độ bắt có thể thay đổi giữa các bệnh nhân và giữa các tổn thương, không phải lúc nào cũng rất cao. Kích thước tổn thương ảnh hưởng rõ đến khả năng phát hiện: tổn thương càng nhỏ thì hiệu ứng thể tích phần (partial volume) càng làm tín hiệu yếu, dễ âm tính giả. Ngoài ra, vì acetate cũng có thể tham gia chuyển hóa năng lượng ở nhiều mô, nên tương phản nền không phải lúc nào cũng tối ưu như các tracer nhắm đích đặc hiệu.
Ưu điểm
Ưu điểm là ¹¹C-acetatephản ánh một nhánh chuyển hóa khác so với với FDG. FDG chủ yếu phản ánh “ăn đường” (chuyển hóa glucose), trong khi acetate phản ánh xu hướng dùng nguyên liệu để tạo lipid và tham gia chuyển hóa năng lượng. Vì vậy, ở một số RCC có bắt FDG kém, acetate có thể vẫn bắt và giúp nhìn thấy tổn thương. Vì vậy, kỹ thuật này có thể giúp hỗ trợ chẩn đoán trong các ca khó.
Hạn chế
Tính khả dụng không cao do ¹¹C có chu kỳ bán rã rất ngắn (khoảng 20 phút), nên thường cần cyclotron và quy trình sản xuất ngay tại chỗ hoặc rất gần nơi chụp. Điều này làm chi phí và logistics tăng, khó triển khai rộng. Về mặt sinh học u, acetate không nhắm một đích phân tử “đặc hiệu cho RCC”, nên độ bắt có thể không đồng nhất và có nguy cơ chồng lấp với các quá trình lành tính có chuyển hóa acetate tương đối cao. Về mặt kỹ thuật, tổn thương nhỏ dễ âm tính giả do hiệu ứng thể tích phần và do chuyển động hô hấp, nên giá trị của xét nghiệm giảm khi mục tiêu là tìm di căn nhỏ. Cuối cùng, vì bằng chứng lâm sàng còn hạn chế và không đồng nhất giữa các nghiên cứu, kỹ thuật này hiện thường được xem là lựa chọn trong hoàn cảnh chọn lọc hơn là công cụ chuẩn thường quy.
PET/CT với Girentuximab gắn đồng vị 89Zr hoặc 124I
Cơ sở phân tử và cơ chế, viết đơn giản nhưng chuẩn thuật ngữ
Girentuximab (còn gọi là cG250) là kháng thể đơn dòng nhận diện đặc hiệu Carbonic anhydrase IX(CAIX), một protein xuyên màng nằm trên bề mặt tế bào. Trong clear cell renalcell carcinoma (ccRCC), CAIX thường tăng biểu hiện rất mạnh vì liên quan trụcVHL–HIF: khi mất chức năng VHL hoặc kích hoạt đường đáp ứng thiếu oxy, Hypoxia-inducible factor (HIF) tăng hoạt tính và thúc đẩy phiên mã CAIX. CAIX giúp tế bào u thích nghi môi trường thiếu oxy bằng cách điều hòa pH ngoại bào và nội bào.
Khi tiêm girentuximab đã gắn đồng vị phóng xạ, kháng thể sẽ lưu hành trong máu, tiếp cận mô u và gắn vào CAIX trên màng tế bào u. PET/CT ghi nhận phân bố phóng xạ theo thời gian, nhờ đó “đánh dấu” được các ổ u giàu CAIX, đặc biệt là ccRCC.
Khác biệt thực hành giữa hai đồng vị:
¹²⁴I-girentuximab: iod là chất có thể bị “tách” khỏi kháng thể (deiodination) trong cơ thể, làm tăng phóng xạ tự do có thể đi tới tuyến giáp; vì vậy thường cần lưu ý nền sinh lý và kiểm soát iod tự do.
⁸⁹Zr-girentuximab: zirconium-89 là đồng vị “phù hợp” với dược động học kháng thể vì chu kỳ bán rã dài và thường cho phép chụp muộn khi nền máu giảm; tuy nhiên nếu ⁸⁹Zr tự do xuất hiện có thể gây bắt nền không mong muốn ở một số mô (vì vậy chất lượng gắn chelate và kiểm soát tạp rất quan trọng).
Vai trò trong RCC
- Định danh không xâm lấn ccRCC trước can thiệp: hỗ trợ phân loại khối u thận theo kiểu mô học “khả dĩ” khi CT/MRI chưa đủ chắc chắn, từ đó hỗ trợ lựa chọn chiến lược theo dõi, sinh thiết hay phẫu thuật.
- Đánh giá toàn thân: có thể phát hiện thêm các ổ tổn thương ccRCC ngoài thận hoặc di căn trong một số bối cảnh, giúp hoàn thiện bức tranh bệnh học trước điều trị.
- Hỗ trợ chọn vị trí sinh thiết: khi tổn thương không đồng nhất, vùng bắt CAIX mạnh có thể là vùng giàu tế bào u sống, tăng khả năng sinh thiết trúng đích.
Đặc điểm hình ảnh của RCC
- ccRCC: thường thấy tăng bắt rõ tại khối u nguyên phát và/hoặc ổ di căn, thường tương phản tốt khi chụp ở thời điểm muộn sau tiêm (khi nền máu đã giảm). Mức bắt có thể không đồng nhất nếu khối u có hoại tử, xuất huyết hoặc vùng giảm tế bào.
- Non-clearcell RCC (papillary, chromophobe và các subtype khác): thường bắt thấp hơn hoặc âm tính tương đối do CAIX biểu hiện thấp hơn.
- Nền sinh lý: có thể còn hoạt độ trong lòng mạch (blood pool) ở giai đoạn sớm; một số cơ quan hệ võng nội mô có thể có nền; với ¹²⁴I cần chú ý khả năng có hoạt độ iốt tự do (tuyến giáp) nếu kiểm soát không tốt.
Ưu điểm
- Tính “nhắm trúng đích” theo sinh học u: nhắm CAIX giúp hướng tới một dấu ấn rất điển hình của ccRCC, vì vậy có giá trị như một phương tiện “gợi ý mô học” bằng hình ảnh, điều mà CT/MRI thường khó làm chắc chắn.
- Tăng độ tin cậy chẩn đoán trong khối u thận khó phân loại: đặc biệt khi mục tiêu là nhận diện ccRCC để quyết định mức độ can thiệp.
- Đánh giá toàn thân bằng một lần chụp: hỗ trợ khảo sát tổn thương ngoài thận trong các tình huống chọn lọc.
Hạn chế
- Dược động học kháng thể chậm: kháng thể lớn, cần thời gian để rời khỏi máu và gắn đích tối ưu, nên thường phải chụp muộn; điều này làm quy trình kéo dài, kém thuận tiện hơn so với các tracer phân tử nhỏ.
- Không phải mọi ccRCC đều biểu hiện CAIX cao: ccRCC có thể CAIX thấp hoặc không đồng nhất,gây âm tính giả, nhất là khối u có hoại tử nhiều hoặc biến thiên sinh học.
- Độ đặc hiệu sinh học không tuyệt đối cho mọi tình huống: CAIX cũng liên quan đáp ứng thiếu oxy và có thể xuất hiện ở một số mô hoặc u khác trong các mức độ khác nhau, tạo khả năng dương tính giả tùy bối cảnh.
- Yêu cầu sản xuất và kiểm soát chất lượng phức tạp: đặc biệt với ⁸⁹Zr (chelation) và ¹²⁴I (ổn định iốt gắn), kéo theo hạn chế về tính sẵn có, chi phí và chuẩn hóa liên cơ sở.
- Phơi nhiễm phóng xạ và tối ưu thời điểm chụp: đồng vị chu kỳ bán rã dài và chụp muộn làm tổng phơi nhiễm có thể cao hơn một số tracer khác, cần cân nhắc lợi ích–nguy cơ trong chỉ định.
PET/CT với PSMA gắn đồng vị 68Ga hoặc 18F
Cơ sở phân tử và cơ chế
PSMA (prostatespecific membrane antigen) là một glycoprotein xuyên màng, còn được gọi là glutamate carboxypeptidase II. Trong ung thư tiền liệt tuyến, PSMA thường biểu hiện mạnh trên bề mặt tế bào u, nhưng trong ung thư biểu mô tế bào thận, PSMA thường liên quan chủ yếu đến tế bào nội mô của tân mạch quanh u (tumorassociated neovasculature), đặc biệt hay gặp ở thể clear cell. Khi tiêm chất gắn PSMA có đánh dấu phóng xạ (thường là ligand nhỏ như [⁶⁸Ga]-PSMA-11,[⁶⁸Ga]-PSMA-617 hoặc các chất gắn [¹⁸F] như DCFPyL), phân tử này sẽ gắn vào vùng ngoài tế bào của PSMA trên bề mặt nội mô tân mạch hoặc đôi khi trên tế bào u, sau đó có thể được nội hóa. PET ghi nhận tín hiệu phóng xạ tập trung tại các vùng giàu PSMA, còn CT cung cấp định vị giải phẫu, nhờ đó tạo bản đồ toàn thân của các ổ tổn thương giàu tân mạch biểu hiện PSMA.
Vai trò trong RCC
Trong RCC, PSMA PET/CT được xem là kỹ thuật hình ảnh phân tử có giá trị nhất ở bối cảnh đánh giá giai đoạn và đặc biệt là phát hiện di căn hoặc tái phát khi hình ảnh học hình thái chưa đủ chắc chắn. Lý do là di căn RCC, nhất là clear cell RCC, thường có tân mạch phong phú và có thể biểu hiện PSMA ở nội mô mạch máu, tạo tương phản tốt để phát hiện tổn thương nhỏ ở hạch, xương hoặc mô mềm. Nhiều tổng quan hệ thống và phân tích gộp cho thấy hiệu năng tốt cho phát hiện di căn và tái phát, đồng thời có thể làm thay đổi quyết định điều trị ở một tỷ lệ bệnh nhân có bệnh tiến triển hoặc nghi ngờ tái phát.
Đặc điểm hình ảnh của RCC với PSMA PET/CT
Trên PSMA PET/CT, tổn thương RCC thường biểu hiện như các ổ tăng bắt thuốc mức độ thay đổi, trong đó di căn thường rõ hơn khối u nguyên phát. Một điểm thực hành quan trọng là đánh giá khối u thận nguyên phát có thể khó hơn so với đánh giá di căn vì thận có bắt nền sinh lý và các chất gắn PSMA thường thải qua đường niệu, làm hoạt độ ở nhu mô thận và hệ thu thập nước tiểu cao, giảm tỷ lệ u trên nền và làm tăng nguy cơ bỏ sót hoặc đánh giá thấp tổn thương tại thận. Ngoài ra, mức bắt thuốc có thể không đồng nhất trong cùng một khối u do hoại tử, xuất huyết hoặc khác biệt sinh học giữa các vùng u, nên đôi khi thấy dạng bắt không đồng đều thay vì “bắt đồng nhất” toàn khối.
Ưu điểm
PSMA PET/CT cung cấp đánh giá toàn thân dựa trên sinh học tân mạch u, nhờ đó có thể phát hiện thêm tổn thương di căn so với CT/MRI trong một số tình huống, đặc biệt ở clear cell RCC vốn giàu tân mạch. Về mặt đồng vị, ⁶⁸Ga có lợi thế là có thể lấy từ generator nên thuận tiện ở nơi không có cyclotron, trong khi ¹⁸F thường cho hình ảnh có độ phân giải tiềm năng tốt hơn do năng lượng positron thấp hơn và có thể sản xuất tập trung, phân phối xa nhờ chu kỳ bán rã dài hơn; tuy nhiên lựa chọn thực tế còn phụ thuộc hệ thống cung ứng, quy trình và từng tracer cụ thể.
Hạn chế
PSMA không phải đích “đặc hiệu cho RCC”, mức biểu hiện thay đổi theo subtype và theo từng bệnh nhân,nên có thể âm tính giả, nhất là ở non-clear cell RCC; đồng thời bắt nền ở thận và bài tiết qua nước tiểu làm giảm giá trị khi mục tiêu chính là mô tả khối u nguyên phát. Ngoài ra, vì PSMA liên quan tân mạch, một số tổn thương không phải RCC nhưng có tân mạch hoặc một số quá trình lành tính có thể gây dương tính giả tùy bối cảnh, nên kết quả cần diễn giải cùng CT/MRI và lâm sàng.
Xét nghiệm máu
Mặc dù các xét nghiệm máu không thể chẩn đoán trực tiếp ung thư biểu mô tế bào thận, nhưng có vai trò quan trọng để đánh giá chức năng thận, phát hiện biến chứng và hỗ trợ theo dõi bệnh.
Tổng phân tích tế bào máu
Số lượng hồng cầu:
- Thiếu máu: Do chảy máu trong hoặc do bệnh mãn tính.
- Đa hồng cầu: Ngược lại, một số khối u thận tiết ra hormone erythropoietin (EPO) khiến cơ thể sản xuất quá nhiều hồng cầu.
Số lượng bạch cầu hoặc tiểu cầu cao: Có thể là dấu hiệu của phản ứng viêm do ung thư.
2. Xét nghiệm sinh hóa
Một số chất chỉ điểm khối u (tumor marker): CEA, BTA (bladder tumor antigen) tăng cao trong một số trường hợp. Có thể sử dụng để theo dõi đáp ứng điều trị, phát hiện bệnh tái phát, di căn xa. Tuy nhiên, không thay thế được vai trò của CT trong theo dõi ung thư biểu mô tế bào thận.
Ure, Creatinine: Dùng để tính toán Mức lọc cầu thận (GFR). Chỉ số đánh giá thận còn lại khi phải phẫu thuật cắt bỏ một bên thận.
Canxi máu: tăng canxi huyết có thể là dấu hiệu cho thấy ung thư đã di căn vào xương hoặc do khối u tiết ra các loại hormone làm rối loạn chuyển hóa canxi.
Men gan (ALT, AST, ALP): Nếu men gan tăng cao, nghi ngờ ung thư đã di căn đến gan hoặc do Hội chứng Stauffer (một biến chứng hiếm gặp của ung thư thận gây rối loạn chức năng gan dù chưa di căn tới gan).
Lactate Dehydrogenase (LDH): Nồng độ LDH cao thường liên quan đến các khối u phát triển nhanh và giúp tiên lượng tình trạng bệnh.
Tốc độ lắng máu (ESR) và CRP: Các chỉ số viêm này thường tăng cao ở bệnh nhân bị ung thư thận giai đoạn tiến triển.
Chẩn đoán
Chẩn đoán xác định
Dựa vào triệu chứng lâm sàng, xét nghiệm cận lâm sàng, trong đó chẩn đoán mô bệnh học mang tính chất quyết định.
Chẩn đoán phân biệt
Chẩn đoán phân biệt rộng,về cơ bản bao gồm tất cả các khối u thận, đặc biệt là các u thận khác, và thường gặp nhất gồm:
Các khối u thận
- U tuyến thận:nên được xem là các ung thư biểu mô tế bào thận giai đoạn sớm, kích thước nhỏ.
- U oncocytoma thận: đặc biệt cần lưu ý trong chẩn đoán phân biệt với typ chromophobe.
- U cơ mỡ mạch (angiomyolipoma, AML): thường có thành phần mỡ lớn; tuy nhiên khoảng 4–5% trường hợp có rất ít hoặc không có mỡ.
- Di căn thận.
- U lympho thận.
- U xơ đơn độc của thận: hiếm gặp.
- U thận dạng nang đa ổ: không thể phân biệt với ung thư biểu mô tế bào thận tế bào sáng dạng nang đa ổ.
Các giả u thận
- Nang thận xuất huyết hoặc nang thận phức tạp: đánh giá theo phân loại Bosniak.
- Áp xe thận, viêm thận – bể thận, viêm thận thùy khu trú.
- Nhồi máu thận.
- Trụ Bertin phì đại.
Các tổn thương lan rộng từ cơ quan lân cận
- Các khối u kế cận xân lấn trực tiếp.
- Ung thư biểu mô niệu mạc của bể thận.
- Các u tuyến thượng thận.
- Các khối u sau phúc mạc.
Chẩn đoán giai đoạn
Phân giai đoạn của ung thư biểu mô tế bào thận dựa trên bảng phân loại TNM theo Ủy ban liên hiệp ung thư Hoa Kỳ (American Joint Committee on Cancer – AJCC) phiên bản 8 hướng dẫn.
Bảng 4. Phân loại TNM trong ung thư biểu mô tế bào thận
Bảng 5. Chẩn đoán giai đoạn ung thư biểu mô tế bào thận theo TNM
Phân tầng nguy cơ bệnh
Theo hướng dẫn của Hiệp hội Cơ sở dữ liệu quốc tế về Ung thư biểu mô tế bào thận di căn (International Metastatic Renal Cell Carcinoma Database Consortium - IMDC), dựa vào các tiêu chí lâm sàng, cận lâm sàng, thời gian, có thể đánh giá tiên lượng và nguy cơ của ung thư biểu mô tế bào thận thành 3 cấp độ nguy cơ, từ tốt đến xấu, bao gồm: Favorable (thuận lợi– tốt); Intermediate (nguy cơ trung gian – vừa); Poor (nguy cơ cao – xấu). Từ đó, hướng tới cân nhắc về các phương pháp điều trị, theo dõi phù hợp đối với từng bệnh nhân.
Phân tầng nguy cơ
Nguy cơ tốt (Favorable): Điểm 0
Nguy cơ trung gian (Intermediate): Điểm 1–2
Nguy cơ xấu (Poor): Điểm ≥ 3
Bảng 6. Thang điểm Karnofsky
Điều trị ung thư biểu mô tế bào thận
Điều trị ung thư thận dựa vào: (1) giai đoạn bệnh: khu trú/ lan tràn; (2) thể mô bệnh học: clear cell RCC hay non-clear cell RCC; (3) phân tầng nguy cơ tái phát di căn (theo bảng phân loại IMDC).
Điều trị ung thư biểu mô tế bào thận ở mỗi giai đoạn khác nhau tương ứng với mỗi mục tiêu khác nhau: triệt căn hay chữa khỏi hoàn toàn (giai đoạn khu trú); kiểm soát bệnh kéo dài và bảo tồn chức năng (giai đoạn tiến triển tại chỗ) → kéo dài sống còn, giảm triệu chứng, tối ưu chất lượng sống (giai đoạn tiến triển muộn,ung thư di căn xa).
Phương án điều trị cần tiếp cận đa chuyên khoa (tiết niệu, ung bướu, chẩn đoán hình ảnh, xạ trị, giải phẫu bệnh) và cá thể hóa theo chức năng thận, bệnh kèm theo,nguy cơ độc tính.
Điều trị ung thư biểu mô tế bào thận giai đoạn khu trú:
Có 3 phương pháp, chiến lược trong tiếp cận điều trị ung thư biểu mô tế bào thận giai đoạn khu trú, bao gồm:
Phẫu thuật là tiêu chuẩn vàng cho RCC giai đoạn khu trú. Gồm có:
+Phẫu thuật cắt thận triệt căn (Radical Nephrectomy/ RN)
+Phẫu thuật cắt thận bán phần, chỉ cắt phần khối u và một phần nhu mô thận xung quanh, bảo tồn nhu mô thận lành còn lại (Partial Nephrectomy/ PN)
Đốt u tại chỗ là lựa chọn thay thế khi phẫu thuật nguy cơ cao hoặc bệnh nhân không đủ điều kiện.
Theo dõi chủ động là chiến lược theo dõi khi khối u nhỏ và ít tiến triển hoặc bệnh nhân không muốn/không thể can thiệp ngay.
Ngoài ra với thể mô bệnh học đặc biệt, liên quan đến hội chứng von Hippel–Lindau, cần tiếp cận theo hướng kết hợp điều trị toàn thân kể cả khi bệnh còn đang ở giai đoạn khu trú.
Theo hướng dẫn của ESMO năm 2024, phương án lựa chọn trong điều trị ung thư biểu mô tế bào thận như sau:
Khối u T1 (≤ 7 cm)
Cắt thận bán phần (Partialnephrectomy, PN)
Cơ chế điều trị của phẫu thuật cắt thận bán phần
Hình 15. Phẫu thuật cắt thận bán phần
Phẫu thuật cắt thận bán phần là phương pháp điều trị tại chỗ nhằm loại bỏ triệt để khối u nguyên phát trong khi vẫn bảo tồn tối đa nhu mô thận lành. Về mặt ung thư học, việc cắt bỏ hoàn toàn khối u cùng với diện cắt âm tính giúp loại trừ nguồn tế bào ác tính đang tăng sinh, qua đó đạt được kiểm soát tại chỗ hiệu quả đối với các khối u thận khu trú, đặc biệt ở giai đoạn T1. Nhiều dữ liệu cho thấy hiệu quả kiểm soát ung thư của cắt thận bán phần tương đương với cắt thận toàn bộ trong các trường hợp được chỉ định phù hợp, phản ánh việc chấm dứt quá trình tiến triển của ung thư ngay tại ổ nguyên phát trước khi xảy ra di căn có ý nghĩa lâm sàng.
Từ góc độ sinh lý bệnh học, điểm khác biệt quan trọng của cắt thận bán phần so với cắt thận toàn bộ là khả năng bảo tồn nephron. Việc duy trì nhu mô thận chức năng giúp bảo tồn mức lọc cầu thận, làm giảm nguy cơ xuất hiện hoặc tiến triển bệnh thận mạn sau phẫu thuật. Điều này có ý nghĩa sinh học và lâm sàng lâu dài, bởi suy thận mạn liên quan đến tình trạng viêm hệ thống, rối loạn chức năng nội mô và tăng nguy cơ tim mạch, những yếu tố có thể ảnh hưởng bất lợi đến sống còn toàn bộ của bệnh nhân ung thư, dù không trực tiếp liên quan đến kiểm soát khối u.
Ở mức độ vi môi trường u, khối u thận là nguồn tiết nhiều yếu tố gây ức chế miễn dịch và thúc đẩy tân sinh mạch, bao gồm VEGF và các cytokine điều hòa âm tính đáp ứng miễn dịch. Việc loại bỏ khối u nguyên phát làm giảm gánh nặng các tín hiệu sinh học bất lợi này, từ đó có thể góp phần cải thiện trạng thái miễn dịch toàn thân. Mặc dù cắt thận bán phần không phải là một liệu pháp miễn dịch, việc loại bỏ ổ điều hòa miễn dịch bệnh lý của khối u được xem là tạo nền sinh học thuận lợi cho hệ miễn dịch tự nhiên và cho các chiến lược điều trị bổ trợ khi cần thiết.
Ngoài ra, cắt thận bán phần làm giảm tổng khối lượng tế bào ung thư trong cơ thể, qua đó làm giảm xác suất tồn tại và tiến hóa của các dòng tế bào ác tính có tiềm năng xâm lấn hoặc kháng trị. Trong ung thư học,việc giảm gánh nặng u sớm được coi là một yếu tố quan trọng giúp làm chậm tiến trình tiến triển bệnh và cải thiện kết cục dài hạn ở những bệnh nhân được chẩn đoán ở giai đoạn sớm.
Tóm lại, cơ chế điều trị của phẫu thuật cắt thận bán phần dựa trên sự kết hợp giữa kiểm soát ung thư tại chỗ bằng cách loại bỏ hoàn toàn khối u nguyên phát và bảo tồn tối đa chức năng thận. Cách tiếp cận này cho phép đạt hiệu quả ung thư học tương đương cắt thận toàn bộ ở các trường hợp phù hợp, đồng thời hạn chế các hậu quả sinh học và lâm sàng lâu dài của mất chức năng thận, từ đó góp phần cải thiện sống còn toàn bộ của bệnh nhân.
Khuyến cáo
PN là lựa chọn ưu tiên hàng đầu đối với các khối u khu trú trong thận có kích thước ≤ 7 cm (chỉ định chủ động). Khuyến cáo này dựa trên một tổng quan hệ thống gồm nhiều nghiên cứu hồi cứu và một thử nghiệm lâm sàng ngẫu nhiên có đối chứng so sánh giữa cắt thận triệt căn (radical nephrectomy, RN) và cắt thận bán phần ở các bệnh nhân có u thận đơn độc giai đoạn T1a–T1b, N0, M0 (< 5 cm) và chức năng thận bên đối diện bình thường.Các dữ liệu này cho thấy cắt thận bán phần giúp bảo tồn chức năng thận tốt hơn một cách có ý nghĩa thống kê so với cắt thận triệt căn.
Cắt thận bán phần có thể được thực hiện bằng phẫu thuật mở, nội soi ổ bụng hoặc nội soi có hỗ trợ robot. Trong trường hợp không thể thực hiện PN do hạn chế kỹ thuật, cắt thận triệt căn bằng nội soi thường hoặc robot được khuyến cáo.
Chiến lược bảo tồn nephron (nephron-sparing), bao gồm PN, được xem là tiêu chuẩn điều trị ở các bệnh nhân có suy giảm chức năng thận, chỉ còn một thận, hoặc u thận hai bên, không phụ thuộc vào kích thước khối u (chỉ định bắt buộc).
Sinh thiết khối u thận trước phẫu thuật đối với các khối u T1a trên lâm sàng được khuyến cáo, vì lên đến 30% trong số này là tổn thương lành tính và có thể không cần can thiệp điều trị; tuy nhiên, chưa có sự đồng thuận tuyệt đối về chỉ định này.
Các phương pháp điều trị không phẫu thuật
Đốt sóng cao tần (radiofrequency ablation, RFA), đốt vi sóng (microwave ablation) và đốt u bằng nhiệt lạnh (cryoablation, CA), xạ trị định vị thân (stereotactic body radiotherapy, SBRT) là các lựa chọn không phẫu thuật, đặc biệt phù hợp với các khối u vỏ thận kích thước nhỏ. Những phương pháp này đặc biệt thích hợp cho bệnh nhân:
- thể trạng yếu hoặc nguy cơ phẫu thuật cao,
- chỉ còn một thận,
- suy giảm chức năng thận,
- RCC di truyền,
- nhiều khối u hai bên,
- từ chối phẫu thuật.
Trong các trường hợp này, sinh thiết trước can thiệp được khuyến cáo để xác nhận tính ác tính và phân nhóm mô bệnh học.
Cơ chế tác dụng của 3 phương pháp đốt u (ablation)
Đốt sóng cao tần (radiofrequency ablation, RFA) là phương pháp điều trị tại chỗ dựa trên cơ chế gây hoại tử đông nhiệt của mô u. Dưới hướng dẫn hình ảnh, một điện cực được đưa trực tiếp vào khối u thận và phát ra dòng điện xoay chiều tần số cao, tạo dao động ion trong mô. Dao động này sinh nhiệt tại chỗ, làm nhiệt độ mô tăng lên khoảng 60–100°C, dẫn đến biến tính protein,phá hủy màng tế bào và gây chết tế bào ung thư không hồi phục.
Về mặt ung thư học, RFA tiêu diệt trực tiếp các tế bào ác tính trong vùng được đốt, qua đó đạt được kiểm soát tại chỗ đối với các khối u thận nhỏ, thường là u vỏ thận giai đoạn sớm. Hiệu quả của RFA phụ thuộc vào việc bao phủ đầy đủ toàn bộ thể tích khối u và một vành mô lành xung quanh,nhằm hạn chế nguy cơ tồn tại tế bào ung thư vi thể và tái phát tại chỗ.
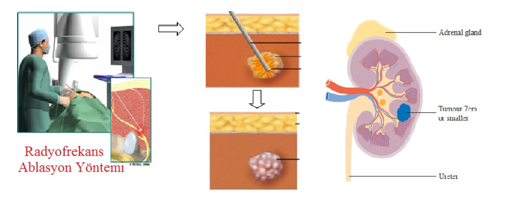
Hình 16. Đốt sóng cao tần điều trị ung thư biểu mô tế bào thận
Đốt vi sóng (microwave ablation) là một phương pháp điều trị tại chỗ dựa trên cơ chế sinh nhiệt trực tiếp từ trường điện từ visóng để tiêu diệt tế bào ung thư. Khi ăng-ten vi sóng được đặt vào khối u dưới hướng dẫn hình ảnh, năng lượng vi sóng làm các phân tử nước trong mô dao động nhanh theo chiều của trường điện từ, tạo ra nhiệt độ cao trong mô u. Nhiệt độ thường vượt quá ngưỡng gây hoại tử đông, dẫn đến biến tính protein, phá hủy cấu trúc màng và bào quan, gây chết tế bào ung thư không hồi phục.
Về mặt ung thư học, microwave ablation tạo ra vùng hoại tử lớn và đồng nhất hơn so với đốt sóng cao tần, do cơ chế sinh nhiệt không phụ thuộc vào dẫn điện của mô. Điều này giúp giảm ảnh hưởng của hiện tượng “tản nhiệt theo dòng máu” và cho phép tiêu diệt hiệu quả các khối u thận có kích thước lớn hơn hoặc nằm gần các cấu trúc mạch máu.
Đốt khối u bằng nhiệt lạnh (cryoablation) là phương pháp điều trị tại chỗ dựa trên cơ chế gây chết tế bào bằng chu kỳ đông – rã đông nhanh. Dưới hướng dẫn hình ảnh, các đầu dò lạnh được đưa trực tiếp vào khối u thận và tạo nhiệt độ rất thấp, thường từ −40°C trở xuống. Nhiệt độ này gây hình thành tinh thể băng nội bào và ngoại bào, làm phá vỡ màng tế bào, tổn thương bào quan và dẫn đến chết tế bào ung thư không hồi phục. Chu kỳ rã đông tiếp theo gây phù nề thẩm thấu và tổn thương vi mạch, làm tăng thêm mức độ hoại tửmô.
Về mặt ung thư học, cryoablation tiêu diệt tế bào ác tính trong vùng được làm lạnh, đạt kiểm soát tại chỗ đối với các khối u thận nhỏ, đặc biệt là các u vỏ thận giai đoạn sớm. Một ưu điểm quan trọng của cryoablation là khả năng quan sát trực tiếp “quả cầu băng” trên hình ảnh học trong quá trình thủ thuật, cho phép kiểm soát chính xác phạm vi điều trị và đảm bảo bao phủ toàn bộ khối u cùng một vành mô an toàn xung quanh, từ đó giảm nguy cơ sót tế bào ung thư vi thể.
Hình 17. A. Bệnh nhân nam 53 tuổi, có tiền sử ung thư thực quản, được phát hiện có khối u thận trái kích thước 3,6 cm. Sinh thiết cho thấy ung thư biểu mô tế bào thận, thể nhú type 1. B. Hình ảnh cắt lớp vi tính (CT) trục ngang của thận trong quá trình đốt lạnh cho thấy ba trong bốn kim cryoablation nằm trong khối “quả cầu băng”giảm tỷ trọng. C. Ngay sau hai chu kỳ đông–rã đông, CT có tiêm thuốc cản quang ghi nhận hiện tượng thoát mạch đang hoạt động kèm theo tụ máu sau phúc mạc. Can thiệp nút mạch chọn lọc (không hiển thị) đã được thực hiện khẩn trương do huyết áp bệnh nhân giảm. D. Hình ảnh CT có tiêm thuốc cản quang theo dõi sau 12 tháng cho thấy vùng đốt u, không có bằng chứng bệnh tồn lưu. (Bản quyền: KamranArhar, MD.)
Hình 18. A. Hình ảnh cắt lớp vi tính (CT) trục ngang của thận trong quá trình đốt lạnh một khối u ở cực trên thận trái cho thấy khối “quả cầu băng” đang phát triển (mũi tên) nằm sát đại tràng (dấu sao). B. Thủ thuật hydrodissection (tách mô lân cận bằng dịch) được thực hiện bằng cách bơm 180 mL dịch vô khuẩn có pha một lượng nhỏ thuốc cản quang iod để tách đại tràng ra khỏi khối băng.
Về sinh lý bệnh học, cả 3 phương pháp đốt u phù hợp với ung thư biểu mô tế bào thận ở giai đoạn khu trú, với kích thước nhỏ <3cm. Đốt u là phương pháp bảo tồn nephron vì chỉ phá hủy mô trong vùng đích,giữ lại phần lớn nhu mô thận chức năng. Sự bảo tồn này giúp duy trì mức lọc cầu thận và hạn chế nguy cơ suy thận mạn, đặc biệt quan trọng ở bệnh nhân có thận đơn độc, suy thận nền hoặc u thận hai bên.
Ở mức độ vi môi trường u, tổn thương nhiệt nóng hay nhiệt lạnh gây phá hủy vi mạch trong khối u, làm mất tưới máu và gây thiếu oxy thứ phát, góp phần tăng hiệu quả tiêu diệt tế bào ác tính. Quá trình hoại tử mô cũng có thể giải phóng các kháng nguyên u và kích hoạt phản ứng viêm tại chỗ, tuy nhiên ý nghĩa của đáp ứng miễn dịch toàn thân liên quan đến các phương pháp này hiện vẫn chưa được chứng minh rõ ràng.
Cơ chế tác dụng của xạ trị định vị thân (Steriotactic body radiotherapy - SBRT)
Xạ trị định vị thân (stereotacticbody radiotherapy, SBRT) điều trị ung thư biểu mô tế bào thận bằng cách chiếu một liều tia xạ rất cao, tập trung chính xác vào khối u, trong khi hạn chế tối đa tia xạ đến mô thận lành xung quanh. Năng lượng bức xạ làm tổn thương trực tiếp vật chất di truyền (DNA) của tế bào ung thư và tạo ra các gốc tự do, khiến tếbào mất khả năng phân chia và dẫn đến chết tế bào.
Ngoài tác động trực tiếp lên tế bào ung thư, liều xạ cao của SBRT còn phá hủy các mạch máu nhỏ nuôi khối u. Khi hệ vi mạch trong u bị tổn thương, nguồn cung cấp oxy và dinh dưỡng cho tế bào ung thư bị cắt đứt, gây thiếu máu cục bộ và hoại tử mô u. Cơ chế này đặc biệt quan trọng trong ung thư thận, vốn có tính kháng xạ tương đối khi dùng xạ trị liều thấp kéo dài.
So sánh các phương pháp đốt u và xạ trị với phẫu thuật
Các nghiên cứu tổng quan hệ thống cho thấy tỷ lệ sống còn đặc hiệu theo nguyên nhân lâu dài sau RFA tương đương với cắt thận bán phần, với tỷ lệ di căn thấp, nhưng tỷ lệ tái phát tại chỗ hơi cao hơn so với PN và cryoablation. Tuy nhiên, chất lượng bằng chứng hiện có chưa đủ mạnh để đưa ra kết luận dứt khoát về độc tính và hiệu quả ung thư học của RFA và CA.
Các dữ liệu từ phân tích gộp, cũng như từ nghiên cứu tiến cứu và hồi cứu, ủng hộ hiệu quả và tính an toàn của SBRT, bao gồm cả kết quả dài hạn thuận lợi. Tuy nhiên, cần thêm các thử nghiệm ngẫu nhiên có đối chứng, và SBRT chưa thể được khuyến cáo mạnh mẽ khi chưa có các dữ liệu này.
Theo dõi chủ động (Activesurveillance)
Theo dõi chủ động là một lựa chọn hợp lý cho bệnh nhân có kỳ vọng sống ngắn hoặc khối u thận nhỏ (≤ 4 cm). Thực tế, tốc độ phát triểncủa khối u thận thường chậm, với mức trung bình khoảng 3 mm mỗi năm, và tỷ lệtiến triển thành bệnh di căn chỉ khoảng 1%–2%. Trong mọi trường hợp, cần cóthảo luận đầy đủ giữa bác sĩ và bệnh nhân về lợi ích và nguy cơ của chiến lượcnày.
Khối u T2 (> 7 cm)
Cắt thận triệt căn (Radical Nephrectomy) xâm lấn tối thiểu (nội soi hoặc robot) là lựa chọn ưu tiên. Các phương pháp tiếp cận khác nhiều khả năng mang lại kết quả ung thư học tương đương.
Hình 19. Phẫu thuật cắt thận triệt căn
Mục tiêu của phẫu thuật trong điều trị ung thư biểu mô tế bào thận (renal cell carcinoma, RCC) là cắt bỏ hoàn toàn khối u với ranh giới phẫu thuật an toàn. Trước đây, cắt thận đơn giản từng được sử dụng rộng rãi, nhưng từ năm 1969, Robson và cộng sự đã xác định cắt thận triệt căn (radical nephrectomy, RN) là tiêu chuẩn vàng cho RCC khu trú. Tuy nhiên, RN hiện nay thường chỉ còn là cắt thận toàn bộ (total nephrectomy), vì không còn thực hiện thường quy cắt tuyến thượng thận hay nạo hạch mở rộng như trước.
Ở các khối u lớn, bóc tách ngoài bao thận vẫn được áp dụng do ≥25% khối u xâm lấn mỡ quanh thận. Cắt tuyến thượng thận cùng bên không còn được khuyến cáo, trừ khi nghi ngờ xâm lấn trực tiếp, vì RCC có xu hướng di căn thượng thận hai bên với tỷ lệ tương đương. Nạo hạch mở rộng cũng không mang lại lợi ích trong RCC khu trú, do tỷ lệ hạch âm tính trên lâm sàng nhưng dương tính mô học <5%, dù vai trò của nạo hạch trong bệnh nguy cơ cao vẫn còn tranh cãi.
RN vẫn là lựa chọn quan trọng cho RCC khu trú, đặc biệt là khối u lớn (đa số T2) hoặc các trường hợp T1 không thể bảo tồn nephron. Theo khuyến cáo AUA 2017, RN nên được cân nhắc ở các khối u đặc hoặc nang phức tạp Bosniak 3–4 có nguy cơ ác tính cao, khi khối u phức tạp, thận đối bên bình thường và eGFR sau mổ dự kiến ≥45 ml/phút/1,73 m², đồng thời không có CKD hay protein niệu từ trước.
Về kỹ thuật, phẫu thuật mở thường dành cho bệnh tiến triển tại chỗ hoặc có hạch lớn. Tuy nhiên, cắt thận triệt căn xâm lấn tối thiểu(nội soi hoặc robot) ngày càng được ưu tiên nhờ hồi phục nhanh và ít biến chứng. Nội soi RN phù hợp với khối u ≤10–12 cm, không xâm lấn cơ quan lân cận hay tĩnh mạch lớn; phẫu thuật robot giúp mở rộng thêm chỉ định trong tay phẫu thuật viên kinh nghiệm.
Dù vậy, RN đang giảm vai trò trong điều trị khối uthận nhỏ (small renal masses, SRM) do nguy cơ gây bệnh thận mạn (CKD). Nghiên cứu cho thấy 26% bệnh nhân SRM dù creatinine bình thường đã có CKD giai đoạn 3(eGFR <60) trước mổ. Sau phẫu thuật, tỷ lệ CKD ≥ giai đoạn 3b (eGFR <45)cao hơn rõ rệt ở nhóm RN so với bảo tồn thận (36% so với 5%, P<0,001). Khi phân tầng nguy cơ CKD theo eGFR và protein niệu, 44% bệnh nhân đã có nguy cơ tiến triển từ trung bình trở lên trước RN.
CKD làm tăng rõ rệt nguy cơ tim mạch và tử vong. Nguycơ tử vong tăng lần lượt 1,2; 1,8; 3,2; và 5,9 lần tương ứng với các mức eGFR45–60; 30–45; 15–30; và <15 ml/phút/1,73 m², ngay cả khi đã điều chỉnh các yếu tố gây nhiễu.
Do nhiều khối u thận nhỏ có diễn tiến sinh học lành tính và không ảnh hưởng đến sống còn, cùng với tác động tiêu cực của RN lên chức năng thận, các dữ liệu này đã củng cố vai trò trung tâm của phẫu thuật bảo tồn nephron trong điều trị RCC khu trú hiện nay.
RCC tiến triển tại chỗ (T3 và T4)
Phẫu thuật cắt thận triệt căn
Cắt thận triệt căn bằng phẫu thuật mở vẫn là tiêu chuẩn điều trị cho các khối u T3 và T4 phức tạp, mặc dù phẫu thuật nội soi hoặc robot có thể được cân nhắc trong một số trường hợp chọn lọc.
Vai trò của nạo hạch bạch huyết trong phẫu thuật (Lymphadectomy)
Không khuyến cáo cắt tuyến thượng thận hoặc nạo hạch thường quy khi chụp CT ổ bụng và thăm dò trong mổ không cho thấy bằng chứng xâm lấn tuyến thượng thận hoặc hạch.
Vai trò của nạo hạch mở rộng trong ung thư biểu mô tế bào thận (RCC) hiện vẫn chưa rõ ràng và còn nhiều tranh cãi. Một số nghiên cứu cũ gợi ý có lợi ích sống còn khi nạo hạch trong lúc cắt thận, nhưng thử nghiệm ngẫu nhiên EORTC 30881 lại không chứng minh được lợi ích sống còn rõ ràng. Kết quả này bị hạn chế vì đa số bệnh nhân trong nghiên cứu có nguy cơ di căn hạch thấp, với 81% khối u độ 1–2, 72% bệnh khu trú trong cơ quan, dẫn đến tỷ lệ di căn hạch chỉ 4% ở nhóm được nạo hạch.
Nghiên cứu của Gershman và cộng sự trên 1.797 bệnh nhân RCC không di căn, sử dụng phân tích propensity score để kiểm soát yếu tố gây nhiễu, cho thấy nạo hạch không cải thiện sống còn (HR 1,14; P = 0,23). Khi phân tích theo nguy cơ có hạch pN1, không ghi nhận lợi ích về sống không tái phát, sống còn đặc hiệu ung thư hay sống toàn bộ.
Để giải thích vì sao nạo hạch không cải thiện tiên lượng, nhóm tác giả đã phân tích 138 bệnh nhân RCC N1M0 đã được cắt bỏ hoàn toàn. Kết quả cho thấy tỷ lệ sống không di căn sau 5 năm chỉ 16%, với thời gian trung bình đến tái phát là 4,2 tháng. Chỉ 16/138 bệnh nhân có đáp ứng lâu dài,và nhóm này có đặc điểm khối u ít xâm lấn hơn, gồm giai đoạn thấp, độ mô học thấp, không hoại tử và không biệt hóa dạng sarcomatoid.
Từ các dữ liệu hiện có, nạo hạch ở bệnh nhân có hạch dương tính trên lâm sàng chủ yếu mang ý nghĩa chẩn đoán giai đoạn hơn là điều trị. Do đó, nạo hạch nên được cân nhắc rất thận trọng, chỉ áp dụng cho bệnh nhân trẻ, RCC dạng nhú type 2 liên quan đến FH, hoặc bệnh nhân tham gia thử nghiệm điều trị bổ trợ, vì đa số bệnh nhân không thu được lợi ích sống cònnhưng lại có nguy cơ biến chứng phẫu thuật.
Trong trường hợp hạch vùng có dấu hiệu xâm lấn rõ trên lâm sàng hoặc hình ảnh học, dù thiếu dữ liệu mạnh, nạo hạch vùng vẫn được xem là tiêu chuẩn hiện nay, đặc biệt ở bệnh nhân chưa có di căn xa (M0).
Vai trò của lấy huyết khối u tĩnh mạch trong phẫu thuật
Các bằng chứng về xử trí huyết khối u trong tĩnh mạch chủ yếu dựa trên nghiên cứu hồi cứu. Phẫu thuật lấy huyết khối tĩnh mạch là kỹ thuật khó, đi kèm nguy cơ biến chứng cao. Can thiệp phẫu thuật có thể được cân nhắc, nhưng chưa xác định được phương pháp tối ưu, và kết cục phụ thuộc nhiều vào mức độ lan của huyết khối.
Ung thư biểu mô tế bào thận (RCC) có khả năng đặc biệt xâm lấn hệ tĩnh mạch, tạo huyết khối u lan từ tĩnh mạch thận vào tĩnh mạch chủ dưới (inferior vena cava, IVC), thậm chí tới tâm nhĩ phải. Khoảng 4–10% bệnh nhân RCC đã có huyết khối u tại thời điểm chẩn đoán.
Huyết khối u được phân loại theo mức độ lan vào IVC (minh họa tại hình dưới).
- Mức 0: giới hạn trong tĩnh mạch thận.
- Mức I: lan vào IVC ≤2 cm trên tĩnh mạch thận.
- Mức II: lan >2 cm nhưng còn dưới tĩnh mạch gan.
- Mức III: lan đến hoặc trên tĩnh mạch gan nhưng dưới cơ hoành.
- Mức IV: lan trên cơ hoành, có thể vào tâm nhĩ phải.
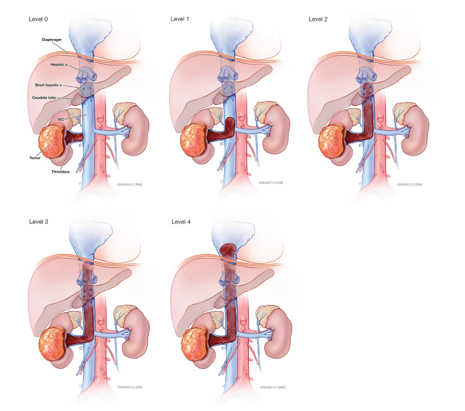
Hình 20. Các mức độ huyết khối u tĩnh mạch trong ung thư biểu mô tế bào thận
Cần nghi ngờ huyết khối u ở bệnh nhân RCC có phù chi dưới mới xuất hiện, giãn tĩnh mạch tinh phải không xẹp khi nằm, giãn tĩnh mạch bụng nông, protein niệu, thuyên tắc phổi, khối trong tâm nhĩ phải, hoặc không thấy thận bị ảnh hưởng trên phim cản quang do tắc tĩnh mạch thận.
Tỷ lệ sống đặc hiệu ung thư sau 5 năm ở bệnh nhân RCC có huyết khối tĩnh mạch dao động 45–70%. Phẫu thuật cắt thận triệt căn kết hợp lấy huyết khối IVC có thể mang lại khả năng chữa khỏi, đáng chú ý là nhiều bệnh nhân không có di căn xa dù huyết khối lan cao.
Giá trị tiên lượng của mức độ huyết khối IVC còn tranh cãi. Phần lớn nghiên cứu cho thấy bệnh nhân huyết khối mức III–IV có nguy cơ tiến triển và tử vong cao hơn. Một số nghiên cứu khác cho rằng xâm lấn IVC nói chung đã là yếu tố tiên lượng xấu, còn di căn hạch và độ ác tính mô học ảnh hưởng mạnh hơn đến sống còn. Dù vậy, bệnh nhân N0M0 ở bất kỳ mức huyết khối nào vẫn có thể được chữa khỏi bằng phẫu thuật.
Dữ liệu hồi cứu cho thấy bệnh nhân mức I–II có thời gian sống trung bình 6,6 năm, cao hơn so với 1,4 năm ở mức III–IV (P = 0,041).Một nghiên cứu đa trung tâm trên 1.774 bệnh nhân ghi nhận tỷ lệ sống đặc hiệu ung thư 5 năm 53,4%, với các yếu tố tiên lượng xấu độc lập gồm: Mô bệnh học dạng nhú (HR 1,62, P <0,05); xâm lấn mỡ (HR 1,49, P <0,01). Phẫu thuật là nền tảng điều trị, với kỹ thuật phụ thuộc mức huyết khối. Huyết khối càng cao,phẫu thuật càng phức tạp, thường cần đội ngũ đa chuyên khoa, đôi khi phải dùng tuần hoàn ngoài cơ thể hoặc ngừng tuần hoàn hạ thân nhiệt. Các yếu tố hình ảnh dự báo cần tái tạo mạch phức tạp gồm: huyết khối bên phải (OR 3,3), đường kính IVC ≥34 mm (OR 4,4), và tắc hoàn toàn IVC (OR 4,6). Nếu không có các yếu tố này, chỉ 2% bệnh nhân cần tái tạo mạch phức tạp.
Tỷ lệ tử vong chu phẫu của RN kết hợp lấy huyết khối IVC là 5–10%. Điều trị tân bổ trợ bằng thuốc nhắm trúng đích không cho thấy hiệu quả rõ ràng trong việc thu nhỏ huyết khối (nghiên cứu lớn nhất 25 bệnh nhân mức III–IV). Một số báo cáo nhỏ gợi ý liệu pháp miễn dịch có thể làm giảm hoặc mất huyết khối. SBRT có thể giúp giảm triệu chứng ở bệnh nhân không thể phẫu thuật do nguy cơ cao hoặc tiên lượng sống hạn chế.
Hiện tại, chưa có vai trò được thiết lập cho điều trị tân bổ trợ (neoadjuvant therapy) trong bối cảnh này.
Các lưu ý đặc biệt ở RCC liên quan hội chứng von Hippel–Lindau (VHL)
Hội chứng von Hippel–Lindau (VHL) là một bệnh di truyền trội trên nhiễm sắc thể thường, hiếm gặp, do đột biến mầm bệnh lý của gen VHL. Khoảng 70% bệnh nhân VHL sẽ phát triển ung thư biểu mô tế bào thận trong suốt cuộc đời.
Trước đây, điều trị RCC ở nhóm bệnh nhân này chủ yếu dựa vào phẫu thuật hoặc các phương pháp đốt u. Tuy nhiên, do khuynh hướng xuất hiện nhiều khối u thận theo thời gian, bệnh nhân thường phải trải qua nhiều lần can thiệp lặp lại.
Belzutifan là một thuốc mới, có cơ chế ức chế yếu tố phiên mã HIF-2α (hypoxia-inducible factor 2 alpha). Trong một thử nghiệm lâm sàng pha II, nhãn mở, đơn nhóm trên 61 bệnh nhân RCC liên quan VHL, belzutifan đạt tỷ lệ đáp ứng toàn bộ (overall response rate, ORR) 64%, đồng thời giảm nhu cầu phải can thiệp điều trị tiếp theo. Thuốc nhìn chung dung nạp tốt và nên được khuyến cáo cho những bệnh nhân chưa cần phẫu thuật ngay lập tức.
Cơ chế tác dụng của Belzutifan
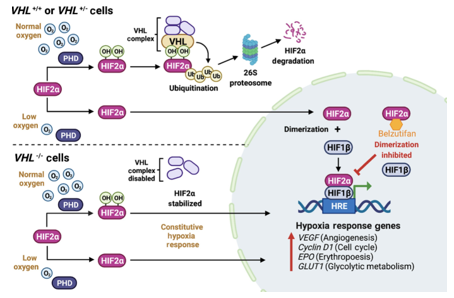
Hình 21. Cơ chế tác động của Belzutifan trong điều trị ung thư biểu mô tế bào thận liên quan đến hội chứng VHL do đột biến
Ở tế bào có VHL bình thường (VHL +/+), khi đủ oxy, enzym prolyl hydroxylase (PHD) hydroxyl hóa HIF-2α. Dạng HIF-2α đã được hydroxyl hóa sẽ được phức hợp VHL nhận diện, gắn ubiquitin và đưa vào proteasome 26S để thoái hóa. Kết quả là HIF-2α không tích lũy, không đi vào nhân và không hoạt hóa các gen đáp ứng thiếu oxy. Khi thiếu oxy, PHD không hoạt động, HIF-2α không bị hydroxyl hóa nên thoát khỏi sự thoái hóa, tích lũy và chuẩn bị tham gia đáp ứng sinh lý với thiếu oxy.
Ở tế bào mất chức năng VHL (VHL −/−), tình huống thay đổi căn bản. Dù oxy bình thường, HIF-2α vẫn không bị nhận diện để thoái hóa vì phức hợp VHL bị vô hiệu. HIF-2α do đó ổn định một cách bất thường, đi vào nhân,dimer hóa với HIF-1β, gắn lên vùng đáp ứng thiếu oxy (HRE) trên DNA và kích hoạt hàng loạt gen liên quan đến sinh ung thư. Các gen tiêu biểu gồm VEGF (tân sinh mạch), Cyclin D1 (chu trình tế bào), EPO (tạo hồng cầu) và GLUT1 (chuyển hóa đường). Hiện tượng này được gọi là đáp ứng thiếu oxy hằng định (constitutive hypoxia response), là trục sinh học trung tâm của ccRCC.
Belzutifan tác động xuống dòng của VHL bằng cách ức chế trực tiếp sự dimer hóa của HIF-2α với HIF-1β. Khi không thể tạo phức dimer, HIF-2α dù đã tích lũy vẫn không hoạt hóa được phiên mã các gen đích, từ đó làmgiảm tân sinh mạch, tăng sinh tế bào và các chương trình chuyển hóa bất lợi cho ung thư. Vì vậy, belzutifan là liệu pháp nhắm trúng đích cơ chế sinh bệnh cốt lõi của ccRCC liên quan đến mất chức năng VHL.
Khuyến cáo của ESMO năm 2024 cũng đưa ra thêm một số khuyến cáo về điều trị bổ trợ bằng liệu pháp toàn thân sau khi điều trị tại chỗ ở các khối u giai đoạn khu trú.
Vai trò của pembrolizumab trong điều trị bổ trợ trong ung thư biểu mô tế bào thận tế bào sáng
Thử nghiệm lâm sàng pha III KEYNOTE-564 đã đánh giá pembrolizumab (17 chu kỳ, liều 200 mg mỗi 3 tuần) so với giả dược trong điều trị bổ trợ sau phẫu thuật ở 994 bệnh nhân ccRCC thuộc nhóm nguy cơ trung bình–cao hoặc nguy cơ cao (theo tiêu chí của nghiên cứu), hoặc bệnh nhân M1 nhưng không còn bằng chứng bệnh (M1 NED).
Sau thời gian theo dõi trung vị 57,2 tháng, pembrolizumab cho thấy cải thiện có ý nghĩa thống kê về sống còn toàn bộ (overall survival, OS) với hazard ratio (HR) 0,62; khoảng tin cậy 95% (CI)0,44–0,87; P = 0,005, đồng thời cải thiện sống không bệnh (disease-freesurvival, DFS) với HR 0,72; 95% CI 0,59–0,87, so với giả dược.
Đây là liệu pháp bổ trợ đầu tiên chứng minh được lợi ích sống còn ở bệnh nhân RCC có khả năng phẫu thuật triệt căn, và hiện được khuyến cáo sử dụng cho bệnh nhân ccRCC nguy cơ trung bình–cao hoặc nguy cơ cao(theo tiêu chí KEYNOTE-564), sau khi lựa chọn bệnh nhân cẩn thận và tư vấn đầy đủ về các tác dụng không mong muốn (adverse events, AEs) có thể xảy ra, cả ngắn hạn và dài hạn.
Khi được chỉ định, điều trị nên bắt đầu trong vòng 12 tuần sau phẫu thuật và kéo dài tối đa 1 năm.
So sánh với các thử nghiệm miễn dịch bổ trợ khác
Lợi ích về DFS và OS ghi nhận trong KEYNOTE-564 khác biệt rõ rệt so với kết quả của các thử nghiệm miễn dịch khác trong bối cảnh bổ trợ, chẳng hạn như: atezolizumab hoặc phối hợp ipilimumab –nivolumab.
Những khác biệt này có thể được giải thích bởi: sự khác nhau về thiết kế thử nghiệm, thời gian điều trị, hoạt tính sinh học của thuốc ức chế điểm kiểm soát miễn dịch (immune checkpoint inhibitors, ICI), hoặc độc tính gia tăng, đặc biệt khi sử dụng ipilimumab. Ngoài ra, dữ liệu biomarker từ các thử nghiệm này vẫn còn cần được phân tích thêm để làm rõ nguyên nhân của sự khác biệt về hiệu quả.
Điều trị đích VEGFR trong bối cảnh bổ trợ: Không được khuyến cáo
Các thuốc nhắm trúng đích thụ thể yếu tố tăng trưởng nội mô mạch máu (VEGFR-targeted therapies) đã cho thấy lợi ích không nhất quán trong các thử nghiệm ngẫu nhiên pha III, do đó không được khuyến cáo trong điều trị bổ trợ RCC.
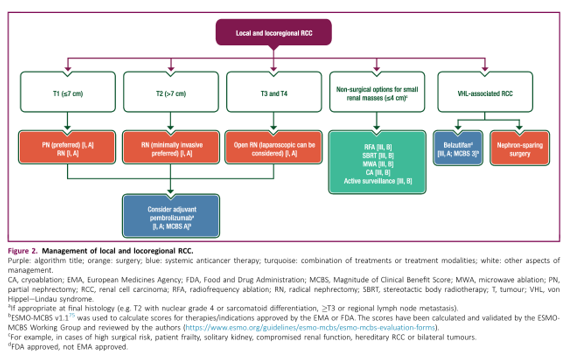
Hình 22. Lưu đồ lựa chọn phương pháp điều trị ung thư biểu mô tế bào thận giai đoạn khu trú theo hướng dẫn của ESMO 2024
Tóm tắt các khuyến cáo điều trị ung thư biểu mô tế bào thận khu trú:
- Phẫu thuật cắt bỏ khối u vẫn là tiêu chuẩn điều trị (standard of care) cho ung thư thận khu trú, với phẫu thuật xâm lấn tối thiểu hoặc mổ mở được lựa chọn tùy theo kích thước và độ phức tạp của khối u. (Mức độ bằng chứng I, mức khuyến cáo A)
- Nhiều chiến lược bảo tồn nephron, từ theo dõi chủ động đến cắt thận bán phần, được khuyến cáo cho khối u thận nhỏ(T1 ≤ 4 cm). (Mức độ bằng chứng III, mức khuyến cáo B)
- Belzutifan có thể giúp tránh hoặc trì hoãn phẫu thuật và có thể được cân nhắc ở bệnh nhân mang đột biến mầm gen VHL và ung thư thận khu trú. (Mức độ bằng chứng III, mức khuyến cáo A; thang điểm lợi ích lâm sàng ESMO-MCBS phiên bản 1.1: 3; đã được FDA phê duyệt nhưng chưa được EMA phê duyệt)
- Pembrolizumab bổ trợ nên được cân nhắc cho bệnh nhân ccRCC có khả năng phẫu thuật thuộc nhóm nguy cơ trung bình–cao hoặc nguy cơ cao (theo tiêu chí KEYNOTE-564), sau khi tư vấn kỹ cho bệnh nhân về các tác dụng phụ dài hạn tiềm tàng. Điều trị nên bắt đầu trong vòng 12 tuần sau phẫu thuật và kéo dài tối đa 1 năm. (Mức độ bằng chứng I,mức khuyến cáo A; ESMO-MCBS v1.1: A)
- Các liệu pháp nhắm trúng đích VEGFR trong điều trị bổ trợ không được khuyến cáo. (Mức độ bằng chứng I, mức khuyến cáo D)
Điều trị RCC ở giai đoạn tiến triển hay di căn xa
Nhìn chung, ở giai đoạn tiến triển hay di căn xa, việc điều trị ung thư biểu mô tế bào thận chủ yếu dựa vào vai trò nổi bật các phương pháp điều trị toàn thân (liệu pháp điều trị đích, điều trị miễn dịch, …), có hoặc không đi kèm với các phương pháp điều trị tại chỗ như phẫu thuật,… Ngoài ra, một số phương pháp mới cũng đang được nghiên cứu, mở ra thêm nhiều triển vọng trong điều trị ung thư biểu mô tế bào thận trong tương lai. Với mỗi thể mô bệnh học, vai trò của mỗi phương pháp điều trị cũng có những sự khác nhau nhất định.
Ung thư biểu mô tế bào thận thể tế bào sáng (clear-cell RCC/ccRCC)
Vai trò của phẫu thuật và các phương pháp điều trị tại chỗ trong ccRCC tiến triển và di căn
Cắt thận giảm khối u ban đầu (upfront cytoreductive nephrectomy, CN) hiện không còn được xem là tiêu chuẩn điều trị ở các bệnh nhân không được chọn lọc, bao gồm:
- bệnh nhân ccRCC nguyên phát không triệu chứng, thuộc nhóm nguy cơ trung gian, và
- tất cả bệnh nhân ccRCC nguyên phát không triệu chứng thuộc nhóm nguy cơ xấu, trong bối cảnh bệnh tiến triển hoặc di căn.
Dựa trên tiêu chí chọn bệnh và phân tích dưới nhóm của thử nghiệm CARMENA, cắt thận giảm khối u vẫn có thể được cân nhắc ở một số bệnh nhân được chọn lọc kỹ, cụ thể là:
- bệnh nhân có gánh nặng di căn thấp, chỉ di căn một cơ quan, kèm khối u nguyên phát lớn,hoặc
- bệnh nhân đã đạt được đáp ứng gần hoàn toàn (near complete response, CR) sau điều trị toàn thân ban đầu.
Ở các bệnh nhân này, sau khi thực hiện CN, có thể cân nhắc theo dõi thay vì tiếp tục điều trị toàn thân, tuy nhiên dữ liệu về kết cục dài hạn trong bối cảnh này còn rất hạn chế.
Cắt bỏ tổn thương di căn (metastasectomy), đốt u bằng nhiệt, xạ phẫu định vị, xạ trị định vị thân (SBRT), xạ trị CyberKnife, và xạ trị phân liều lớn (hypofractionated radiotherapy) có thể được cân nhắc cho một số bệnh nhân chọn lọc có gánh nặng di căn thấp, sau khi được hội chẩn bởi hội đồng đa chuyên. Tuy nhiên, hiện chưa có các thử nghiệm ngẫu nhiên hoặc dữ liệu tiến cứu mạnh để khẳng định chắc chắn lợi ích của các phương pháp này. Trên thực tế, các can thiệp này thường chỉ tập trung vào một vị trí tổn thương đơn lẻ.
Trong nghiên cứu KEYNOTE-564, pembrolizumab cho thấy lợi ích về sống không bệnh (DFS) và sống còn toàn bộ (OS) ở những bệnh nhân M1 nhưng không còn bằng chứng sau cắt bỏ tổn thương di căn. Ngược lại, điều trị toàn thân được xem là cách tiếp cận tối ưu đối với các bệnh nhân tái phát sớm (< 1 năm) sau cắt thận, khiến vai trò của phẫu thuật trong nhóm bệnh nhân này trở nên gây tranh cãi. Do đó, các trường hợp này cần được đánh giá trong khuôn khổ hội chẩn đa chuyên khoa, và phẫu thuật thường được tránh.
Ngoài ra, theo dõi chủ động cũng là một lựa chọn hợp lý đối với những bệnh nhân tái phát sau cắt thận nhưng có diễn tiến chậm, gánh nặng bệnh thấp, và thuộc nhóm nguy cơ thuận lợi theo IMDC.
Vai trò của các phương pháp điều trị toàn thân ccRCC tiến triển, di căn xa.
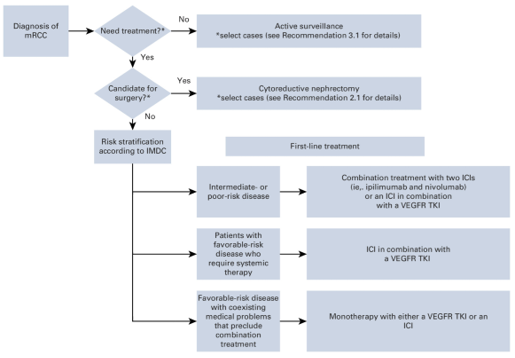
Hình 23. Lưu đồ lựa chọn phương pháp điều trị ung thư biểu mô tế bào thận giai đoạn tiến triển/ di căn xa theo hướng dẫn của ASCO2023.
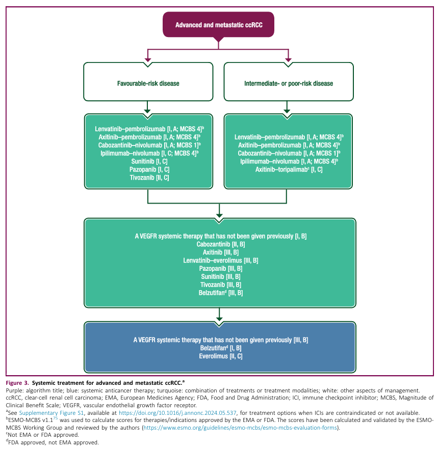
Hình 24. Lưu đồ lựa chọn phương pháp điều trị ung thư biểu mô tế bào thận tiến triển/ di căn xa theo hướng dẫn của ESMO 2024.
Điều trị bước một (First-line treatment)
Điều trị bước một bằng các thuốc ức chế PD-1 phối hợp với liệu pháp nhắm trúng đích thụ thể VEGFR hoặc với ức chế CTLA-4 đã chứng minh cải thiện sống còn toàn bộ (overall survival, OS) ở bệnh nhân ung thư biểu mô tế bào thận tế bào sáng (ccRCC) tiến triển.
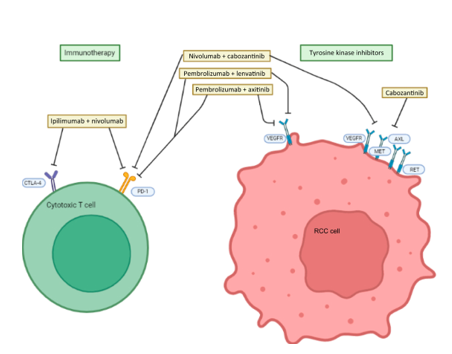
Hình 25. Tổng quan về các liệu pháp điều trị toàn thân ung thư biểu mô tế bào thận, bao gồm điều trị miễn dịch, điều trị đích.
Cơ chế tác dụng của các thuốc ức chế PD-1
Các kháng thể đơn dòng ức chế PD-1 bao gồm: pembrolizumab, nivolumab, toripalimab, …

Hình 26. Cơ chế ức chế và kích hoạt của tế bào T ở điều kiện bình thường
Trong điều kiện bình thường, không có tín hiệu ức chế, sự gắn kết giữa TCR trên tế bào T và phức hợp MHC–peptide trên tế bào trình diện kháng nguyên sẽ khởi phát tín hiệu hoạt hóa qua TCR (signal 1). Nếu đồng thời có tín hiệu đồng kích thích (signal 2), chủ yếu qua trục CD28–B7, tế bào T sẽ tăng sinh, tiết cytokine như interleukin-2, tăng biểu hiện các phân tử gây độc tế bào và trở thành tế bào T hiệu quả có khả năng tiêu diệt tế bào mang kháng nguyên.
Trong bối cảnh có tín hiệu ức chế PD-1/PD-L1, dù TCR vẫn nhận diện đúng kháng nguyên, tín hiệu ức chế từ PD-1 sẽ chặn các đường truyền tín hiệu nội bào của TCR, đặc biệt là các trục liên quan đến phosphoryl hóa kinase và chuyển hóa tế bào. Kết quả là tế bào T không tăng sinh hiệu quả, giảm tiết cytokine, giảm độc tính tế bào và rơi vào trạng thái ức chế chức năng hoặc kiệt quệ (T-cell exhaustion). Đây là cơ chế giúp khối u trốn thoát được về mặt miễn dịch, dù vẫn được nhận diện về mặt kháng nguyên.
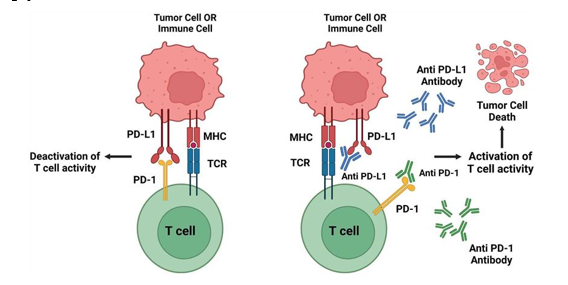
Hình 27. Cơ chế ức chế và kích hoạt tế bào T trong bệnh lý ung thư và cơ chế tác dụng của thuốc ức chế điểm kiểm soát miễn dịch khôi phục hoạt động của tế bào T trong điều trị ung thư
Bên trái, tế bào u (hoặc tế bào miễn dịch trình diện kháng nguyên)biểu hiện PD-L1 trên bề mặt. Khi PD-L1 gắn với PD-1 trên tế bào T, tín hiệu ức chế được truyền vào tế bào T, làm giảm hoặc tắt hoạt tính của tế bào T dù phức hợp MHC–TCR vẫn nhận diện kháng nguyên. Cơ chế này cho phép khối u né tránh đáp ứng miễn dịch, dẫn đến tình trạng “kiệt quệ” chức năng của tế bào T.
Bên phải, các kháng thể kháng PD-1 hoặc kháng PD-L1 chặn sự gắn kết giữa PD-1 và PD-L1. Khi “phanh miễn dịch” này bị tháo bỏ, tín hiệu ức chế không còn, tín hiệu hoạt hóa qua MHC–TCR được khôi phục, tế bào T tăng sinh và tiết cytokine, từ đó tái kích hoạt khả năng tiêu diệt tế bào u. Kết quả cuối cùng là hoạt hóa miễn dịch chống u và tiêu diệt tế bào ung thư.
Một cách đơn giản,có thể hiểu PD-1/PD-L1 là một cơ chế phanh sinh lý của miễn dịch bị khối u lợi dụng; thuốc anti-PD-1/anti-PD-L1 hoạt động bằng cách giải phóng phanh này, giúp hệ miễn dịch nhận diện và tiêu diệt tế bào ung thư hiệu quả hơn.
Cơ chế tác dụng của thuốc ức chế CTLA-4
Các thuốc ức chế CTLA-4 là các kháng thể đơn dòng, gắn trực tiếp với phân tử CTLA-4 để phong bế phân tử này. Các thuốc ức chế CTLA-4 gồm: ipilimumab, tremelimumab,…
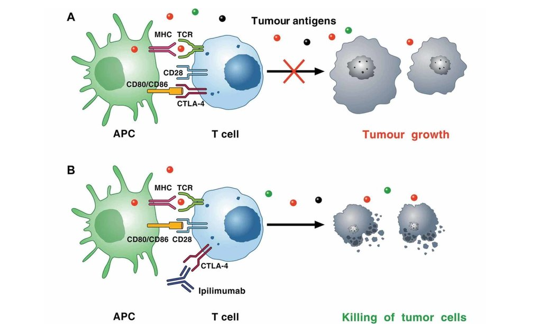
Hình 28. Cơ chế kiểm soát miễn dịch qua CTLA-4 và cơ chế tác dụng của ipilimumab (kháng thể đơn dòng kháng CTLA-4) trong diều trị ung thư.
Tế bào trình diện kháng nguyên (APC) đưa kháng nguyên u cho tế bào T qua phức hợp MHC–TCR. Dù có tín hiệu đồng kích thích CD28–CD80/CD86, sự hiện diện và gắn kết của CTLA-4 với CD80/CD86 (ái lực cao hơn CD28) làm chiếm chỗ tín hiệu đồng kích thích, dẫn đến ức chế hoạt hóa tế bào T. Hệ quả là đáp ứng miễn dịch chống u bị “phanh lại”,các kháng nguyên u không bị loại trừ hiệu quả, và khối u tiếp tục tăng trưởng.
Khi sử dụng ipilimumab (kháng thể kháng CTLA-4), CTLA-4 bị chặn không thể gắn CD80/CD86. Nhờ đó, CD28 tiếp tục nhận tín hiệu đồng kích thích, tín hiệu MHC–TCR được khuếch đại, tế bào T được hoạt hóa bền vững, tăng sinh và thực hiện chức năng gây độc tế bào. Kết quả là tế bào u bị tiêu diệt.
Cơ chế tác dụng của liệu pháp điều trị nhắm trúng đích VEGFR
Vai trò sinh lý của hệ thống VEGF/ VEGFR
VEGFR (vascular endothelial growth factor receptor) là các thụ thể tyrosine kinase giữ vai trò trung tâm trong sinh lý phát triển và duy trì hệ mạch máu. Khi các ligand VEGFgắn vào VEGFR, đặc biệt là VEGFR-2, thụ thể sẽ dimer hóa và hoạt hóa các đường tín hiệu nội bào thúc đẩy tăng sinh, di chuyển và sống còn của tế bào nội mô mạch máu, từ đó hình thành mạch mới. Trong giai đoạn phôi thai, VEGFR cần thiết cho vasculogenesis và angiogenesis, bảo đảm sự phát triển bình thường của hệ tuần hoàn. Ở cơ thể trưởng thành, VEGFR tham gia duy trì tính toàn vẹn mạch máu, điều hòa tính thấm thành mạch, thích ứng với thiếu oxy mô, và tạo mạch trong lành vết thương. Ngoài ra, VEGFR-3 đóng vai trò quan trọng trong phát triển hệ bạch huyết.Nhìn chung, VEGFR là trục tín hiệu sinh lý thiết yếu giúp mô nhận đủ oxy và dinh dưỡng, đồng thời thích nghi với các thay đổi huyết động và chuyển hóa.

Hình 29. Các yếu tố tăng sinh nội mạc mạch VEGF, thụ thể VEGFR và các tác dụng sinh học tương ứng của chúng
VEGF-B và PlGF (placental growth factor) chủ yếu gắn vào VEGFR-1. VEGFR-1 có vai trò sinh lý đặc biệt: nó hoạt động như một “thụ thể mồi” (decoy receptor), điều hòa lượng VEGF tự do, giúp tinh chỉnh đáp ứng tạo mạch, duy trì hằng định chuyển hóa và mạch máu, đồng thời tham gia điều hòa viêm. VEGFR-1 không phải là thụ thể chính thúc đẩy tạo mạch mạnh.
VEGF-A là ligand quan trọng nhất trong sinh lý và bệnh lý, có thể gắn vào VEGFR-1 và VEGFR-2, nhưng tác dụng sinh học chủ yếu thông qua VEGFR-2. Khi VEGF-A hoạt hóa VEGFR-2, tế bào nội mô mạch máu sẽ tăng di chuyển, tăng sinh, sống còn, đồng thời tăng tính thấm thành mạch. Đây là trục chính của angiogenesis (tân sinh mạch) và là nền tảng sinh học của nhiều bệnh lý, trong đó có ung thư biểu mô tế bào thận.
VEGF-C và VEGF-D có thể hoạt hóa VEGFR-2 nhưng đặc biệt gắn vào VEGFR-3. VEGFR-3 là thụ thể chủ chốt của lymphangiogenesis, tức là hình thành và phát triển hệ bạch huyết. Con đường này liên quan mật thiết đến di căn qua hạch bạch huyết của ung thư.
Cơ chế tác dụng của các thuốc TKI kháng VEGFR trong điều trị ung thư biểu mô tế bào thận

Hình 30. Cơ chế và đích tác dụng của các TKI trong điều trị ung thư biểu mô tế bào thận
Ở tế bào ung thư, khi VHL mất chức năng, các yếu tố cảm ứng thiếu oxy HIF-1α và HIF-2α không bị thoái hóa mà tích lũy trong nhân, kết hợp với HIF-1β để hoạt hóa phiên mã. Hệ quả là tăng biểu hiện nhiều gen thúc đẩy tăng trưởng và sống còn của khối u, nổi bật gồm VEGF (tạo mạch), GLUT-1 (tăng thu nhận glucose) và Cyclin-D1 (chu trình tế bào). Các thuốc ức chế HIF-2α (PT2399, PT2385) chặn trực tiếp bước phiên mã phụ thuộc HIF-2α, làm giảm “đầu nguồn” tín hiệu sinh u.
Ở tế bào nội mô mạch máu, VEGF do tế bào u tiết ra gắn vào VEGFR trên nội mô, kích hoạt angiogenesis (tân sinh mạch). Song song, các trục HGF–c-MET và FGF–FGFR cũng thúc đẩy tạo mạch và là cơ chế thoát kháng khi chỉ ức chế VEGF. Do đó, các VEGFR-TKI khác nhau cóphổ đích khác nhau:
- Sunitinib, Pazopanib, Axitinib: ức chế chủ yếu VEGFR, làm giảm tạo mạch.
- Lenvatinib: ức chế VEGFR + FGFR, chặn thêm nhánh FGF.
- Cabozantinib: ức chế VEGFR + c-MET, đặc biệt hữu ích khi u hoạt hóa trục MET.
Các khuyến cáo về lựa chọn thuốc
Thời gian sống còn toàn bộ trung vị ở nhóm bệnh nhân không chọn lọc được điều trị bằng các phác đồ phối hợp nhắm PD-1 hiện nay vượt quá 4 năm.
Các phác đồ phối hợp liệu pháp miễn dịch ức chế PD-1 và liệu pháp đích kháng VEGFR.
Các phối hợp sau được khuyến cáo cho điều trị bước một ccRCC tiến triển, không phụ thuộc vào nhóm nguy cơ IMDC:
- Lenvatinib – pembrolizumab,
- Axitinib – pembrolizumab,
- Cabozantinib – nivolumab.
Các dữ liệu gần đây cũng ủng hộ việc sử dụng axitinib – toripalimab ở bệnh nhân nguy cơ trung gian và nguy cơ xấu, tuy nhiên dữ liệu OS hiện còn chưa hoàn chỉnh. Hiện tại,không có phối hợp nào được xem là ưu tiên hơn rõ rệt, và không khuyến cáo so sánh gián tiếp giữa các thử nghiệm khác nhau. Đáng lưu ý, phối hợp axitinib –avelumab không cho thấy lợi ích OS so với sunitinib.
Phối hợp liệu pháp bộ đôi miễn dịch kháng PD-1 và CTLA-4: Vai trò của ipilimumab – nivolumab
Phối hợp ipilimumab – nivolumab cũng được khuyến cáo như một lựa chọn điều trị tương đương trong điều trị bước một cho bệnh nhân nguy cơ trung gian và nguy cơ xấu theo IMDC, và có thể cân nhắc (với mức khuyến cáo yếu hơn) ở bệnh nhân nguy cơ thuận lợi.
Việc mở rộng khuyến cáo sang nhóm nguy cơ thuận lợi dựa trên:
- hiệu quả cải thiện được ghi nhận trong các phân tích dữ liệu cập nhật gần đây, và
- cơ sở thống kê hợp lệ cho việc đưa nhóm này vào phân tích.
Cụ thể, các tiêu chí đánh giá chính của thử nghiệm pha III CheckMate 214 bao gồm không chỉ nhóm IMDC trung gian và xấu, mà còn cả quần thể theo nguyên tắc “điều trị theo dự định” (intention-to-treat, ITT), trong đó có nhóm nguy cơ thuận lợi. Trong quần thể ITT, OS được cải thiện rõ rệt với HR 0,72; 95% CI 0,62–0,85.
Phân tích dưới nhóm nguy cơ thuận lợi không phải là tiêu chí chính của thử nghiệm. Các kết quả ban đầu không thuận lợi, do đó các khuyến cáo trước đây chỉ giới hạn cho nhóm trung gian và xấu. Tuy nhiên, các kết quả cập nhật gần đây cho thấy triển vọng khả quan hơn, với OS của ipilimumab – nivolumab ở nhóm nguy cơ thuận lợi nằm trong khoảng tương đương với các phối hợp VEGFR – PD-1.
Sau thời gian theo dõi trung vị 67,7 tháng, ipilimumab – nivolumab cho thấy HR OS 0,94 (95%CI 0,65–1,37) ở nhóm nguy cơ thuận lợi. Mặc dù tỷ lệ đáp ứng khách quan (ORR)(30% so với 52%) và sống không tiến triển (progression-free survival, PFS) nghiêng về sunitinib (HR 1,60; 95% CI 1,13–2,26), tỷ lệ đáp ứng hoàn toàn (complete response, CR) (13% so với 6%) và độ bền vững của đáp ứng (59% so với 52% còn đáp ứng sau 5 năm) lại tốt hơn với ipilimumab – nivolumab. Các kết quả theo dõi dài hạn hơn vẫn đang được chờ đợi để xác định xu hướng này có tiếp tục hay không.
Tranh luận về chỉ định ipilimumab – nivolumab ở nhóm nguy cơ thuận lợi:
Nhóm tác giả đã thảo luận rất kỹ về việc khuyến cáo ipilimumab – nivolumab cho bệnh nhân nguy cơ thuận lợi, nhưng không đạt được sự đồng thuận tuyệt đối. Khuyến cáo hiện tại phản ánh đa số (70%) tác giả ủng hộ việc coi ipilimumab – nivolumab là một lựa chọn trong nhóm này.
Lập luận ủng hộ bao gồm: OS cải thiện trong quần thể ITT của CheckMate 214 (bao gồm cả nguy cơ thuận lợi); khả năng đạt đáp ứng hoàn toàn bền vững, vốn hiếm gặp với sunitinib. Lập luận phản đối gồm: không có lợi ích OS rõ ràng trong phân nhóm nguy cơ thuận lợi; PFS và ORR kém hơn sunitinib; chưa có khả năng chọn lọc được bệnh nhân nguy cơ thuận lợi nào sẽ thực sự hưởng lợi từ phối hợp này.
Vấn đề độc tính cũng được thảo luận sâu. Có sự đồng thuận rằng nguy cơ độc tính cấp tính đe dọa tính mạng, cũng như độc tính kéo dài suốt đời, phải được tư vấn rất kỹ cho bệnh nhân nếu cân nhắc sử dụng ipilimumab – nivolumab.
Dù quan điểm khác nhau, các tác giả đều cho rằng phân nhóm nguy cơ IMDC có thể không phản ánh đầy đủ sinh học của bệnh, cũng như đáp ứng với liệu pháp miễn dịch, và do đó cần các biomarker đáng tin cậy hơn để lựa chọn điều trị.
Bảng tổng hợp các thử nghiệm chứng minh lợi ích OS với các liệu pháp: PD-1 – VEGFR; PD-1 – CTLA-4; PD-1 – CTLA-4 – VEGFR, được trình bày trong bảng dưới đây.
Bảng 7. Tóm tắt các thử nghiệm lâm sàng đánh giá hiệu quả của liệu pháp miễn dịch phối hợp kháng VEGFR trong điều trị bước một (first-line) ung thư biểu mô tế bào thận tế bào sáng tiến triển và di căn.
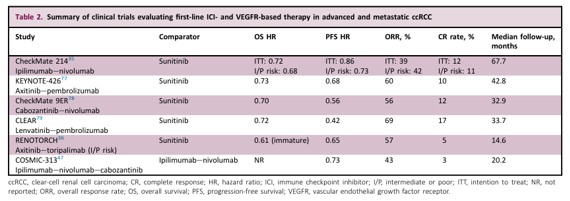
Các tình huống đặc biệt và lựa chọn thay thế
Ở những bệnh nhân chống chỉ định với liệu pháp miễn dịch (ICI) hoặc không tiếp cận được ICI, có thể sử dụng sunitinib, pazopanib hoặc tivozanib. Cabozantinib là một lựa chọn thay thế ở bệnh nhân nguy cơ trung gian và xấu theo IMDC khi không thể dùng liệu pháp nhắm PD-1 bước một.
Theo dõi chủ động
Theo dõi chủ động có thể phù hợp với một số bệnh nhân chọn lọc thuộc nhóm IMDC nguy cơ thuận lợi, có gánh nặng khối u thấp.
Dữ liệu OS ở bệnh nhân IMDC nguy cơ thuận lợi được điều trị bằng phối hợp VEGFR – PD-1 hiện vẫn chưa hoàn chỉnh, và các phác đồ này chưa cho thấy vượt trội rõ rệt so với sunitinib. Tuy vậy, tỷ lệ đáp ứng và PFS tốt hơn vẫn ủng hộ việc sử dụng các phối hợp này trong phân nhóm thăm dò, cỡ mẫu nhỏ.
Do đó, sunitinib, pazopanib và tivozanib vẫn nên được xem là các lựa chọn thay thế hợp lý cho phối hợp VEGFR – PD-1 ở nhóm IMDC nguy cơ thuận lợi, dù mức độ bằng chứng yếu hơn.
Các tình huống sinh học đặc biệt
Liệu pháp phối hợp nhắm PD-1 cho thấy hiệu quả đặc biệt mạnh ở các khối u có đặc điểm sarcomatoid, và do đó được khuyến cáo mạnh mẽ trong nhóm này.
Việc so sánh phác đồ ba thuốc ipilimumab – nivolumab – cabozantinib với ipilimumab – nivolumab ở bệnh nhân chưa điều trị trước đó, thuộc nhóm nguy cơ trung gian hoặc xấu, cho thấy lợi ích rõ rệt về PFS với phác đồ ba thuốc, nhưng đi kèm độc tính tăng cao. Do chưa có dữ liệu OS, phối hợp này chưa được khuyến cáo trong thực hành hiện nay.
Thời gian điều trị tối ưu
Thời gian điều trị tối ưu trong điều trị bước một hiện chưa được xác định rõ ràng.
- Trong CheckMate 214, nivolumab được dùng cho đến khi bệnh tiến triển.
- Trong các phác đồ ICI + VEGFR TKI, thuốc nhắm PD-1 thường được ngừng sau 2 năm.
Việc ngắt quãng điều trị đối với đơn trị liệu nhắm VEGFR không làm giảm hiệu quả điều trị. Lợi ích của việc tiếp tục điều trị nhắm PD-1 quá 2 năm hiện vẫn chưa rõ ràng.
Điều trị bước hai (Second-line treatment)
Các dữ liệu tiến cứu trong bối cảnh điều trị bước hai sau thất bại với liệu pháp nhắm PD-1 ở bước một đã được ghi nhận đối với một số thuốc, bao gồm axitinib, pazopanib, cabozantinib và sunitinib. Tuy nhiên, các kết quả này thường bị ảnh hưởng bởi tính không đồng nhất giữa các thử nghiệm.
Bên cạnh đó,các phân tích hồi cứu và phân tích dưới nhóm mang tính thăm dò cũng đã được báo cáo từ các nghiên cứu sử dụng cabozantinib, tivozanib, phối hợp lenvatinib –everolimus và lenvatinib – pembrolizumab. Trong các nghiên cứu này, tỷ lệ đáp ứng (overall response rate, ORR) dao động khoảng 20%–40%, và các kết cục điều trị phù hợp với kỳ vọng khi áp dụng chiến lược điều trị theo chuỗi (sequencingtherapy).
Do những hạn chế về chất lượng dữ liệu, các thuốc nói trên được khuyến cáo một cách thận trọng.Tuy nhiên, mặc dù có những thiếu sót của các so sánh hồi cứu và gián tiếp, hiệu quả của các thuốc này dường như tương đương với hiệu quả của liệu pháp nhắm VEGFR bước hai trong thời kỳ trước miễn dịch. Vì vậy, chiến lược tiếp tục điều trị theo chuỗi bằng các thuốc nhắm VEGFR vẫn được khuyến cáo mạnh mẽ.
Không khuyến cáo tiếp tục liệu pháp miễn dịch sau thất bại phối hợp PD-1 bước một
Việc sử dụng liệu pháp miễn dịch tiếp theo (ICI rechallenge) sau khi đã điều trị thất bại với phối hợp nhắm PD-1 ở bước một không được khuyến cáo, và có thể gây hại.
Thử nghiệm phaIII CONTACT-03 đã đánh giá atezolizumab (1200 mg truyền tĩnh mạch mỗi 3 tuần) phối hợp với cabozantinib (60 mg uống mỗi ngày) so với cabozantinib đơn trị ở các bệnh nhân tiến triển sau điều trị bằng ICI.
Sau thời gian theo dõi trung vị 15,2 tháng, nghiên cứu không chứng minh được lợi ích về: sống còn toàn bộ (OS): HR 0,94; 95% CI 0,70–1,27; P =0,69 và sống không tiến triển (PFS): HR 1,03; 95% CI 0,83–1,28; P = 0,78 khi sử dụng tái thử miễn dịch (ICI rechallenge). Ngược lại, độc tính tăng lên rõ rệt với liệu pháp miễn dịch bước hai: biến cố bất lợi nghiêm trọng xảy ra ở 48% bệnh nhân dùng atezolizumab – cabozantinib, so với 33% ở nhóm cabozantinib đơn trị.
Cần lưu ý rằng vai trò của việc sử dụng liên tiếp hai liệu pháp miễn dịch trong trường hợp khoảngthời gian không bệnh kéo dài sau điều trị trước đó chưa được nghiên cứu đầy đủ.Một số thử nghiệm khác đang tiếp tục khảo sát vấn đề này.
Cabozantinib: lựa chọn ưu tiên ở bước hai
Tỷ lệ đáp ứng ấn tượng (ORR 40,9%) và PFS trung vị 10,8 tháng ghi nhận ở nhánh cabozantinib đơn trị trong CONTACT-03 khiến cabozantinib bước hai trở thành một lựa chọn rất hấp dẫn.
Kết quả tương tự cũng được ghi nhận trong nghiên cứu pha III CANTATA, đánh giá telaglenastat – cabozantinib so với cabozantinib đơn trị, trong đó nhánh cabozantinib đạt: ORR28%, PFS trung vị 9,3 tháng.
Đáng chú ý, 100% bệnh nhân trong CONTACT-03 và 62% bệnh nhân trong CANTATA đã được điều trị bằng ICI trước đó. Những kết quả này củng cố vai trò của cabozantinib như thuốc nhắm VEGFR được ưu tiên ở bước hai, nếu chưa được sử dụng ở bước một.
Vai trò của belzutifan ở bước hai
Thử nghiệm phaIII LITESPARK-005, so sánh belzutifan với everolimus ở bệnh nhân ccRCC đã được điều trị trước, bao gồm một nhóm nhỏ bệnh nhân (13%) chỉ mới trải qua một dòng điều trị trước đó.
Dựa trên lợi ích PFS vượt trội so với everolimus trong toàn bộ quần thể nghiên cứu, belzutifan có thể được xem là một lựa chọn điều trị bước hai sau tiến triển trên phối hợp VEGFR – PD-1. Tuy nhiên, mức độ khuyến cáo yếu hơn so với vai trò của belzutifan ở bước ba, và cần cân nhắc rằng các lựa chọn khác như cabozantinib có thể phù hợp hơn trong bối cảnh này.
Điều trị bước ba (Third-line treatment)
Ở bệnh nhân ccRCC đã được điều trị nhiều dòng, bao gồm cả ICI và liệu pháp nhắm VEGFR, belzutifan cho thấy lợi ích rõ rệt về PFS so với everolimus, với HR PFS 0,75;95% CI 0,63–0,90.
Ngoài ra, ORR cao hơn đáng kể cũng được ghi nhận với belzutifan (23% so với 4%). Phân tích OS tạm thời không cho thấy lợi ích sống còn (HR 0,88; 95% CI 0,73–1,07), tuy nhiênđộc tính và chất lượng sống đều nghiêng về belzutifan.
Do đó, belzutifan nên được ưu tiên sử dụng thay cho everolimus trong bối cảnh này. Tiếp tục điều trị theo chuỗi bằng các thuốc nhắm VEGFR là một lựa chọn thay thế hợp lý cho belzutifan.
Nguyên tắc chung về điều trị các dòng sau
Nhiều khả năng việc sử dụng tuần tự các thuốc nhắm trúng đích khác nhau đã được phê duyệt trong RCC tiến triển mang lại lợi ích, tương tự như kinh nghiệm trong thời kỳ trước miễn dịch.
Ngược lại, tái thử liệu pháp miễn dịch (ICI rechallenge) hiện chưa có bằng chứng thuyết phục và không nên được xem là lựa chọn chuẩn trong thực hành lâm sàng.
Điều trị ung thư biểu mô tế bào thận thể nhú (papillary RCC, pRCC) tiến triển và di căn
Phẫu thuật
Vai trò của cắt thận giảm khối u (cytoreductive nephrectomy, CN) và các kỹ thuật phẫu thuật khác trong pRCC di căn hiện chưa được xác định rõ ràng.
Phẫu thuật có thể phù hợp ở một số bệnh nhân nguy cơ trung gian, tuy nhiên ở bệnh nhân nguy cơ xấu, lợi ích rất hạn chế và nên tránh phẫu thuật trong bối cảnh này.
Hiện chưa có sự đồng thuận về việc xác định chính xác nhóm bệnh nhân nào sẽ hưởng lợi từ phẫu thuật.
Điều trị bước một
Mặc dù đã có nhiều tiến bộ trong điều trị ccRCC, các nghiên cứu chất lượng cao hướng dẫn điều trị các thể không phải tế bào sáng, bao gồm pRCC, vẫn còn rất hạn chế.
Một thuật toán điều trị toàn thân cho pRCC tiến triển và di căn được trình bày trong Hình 4.
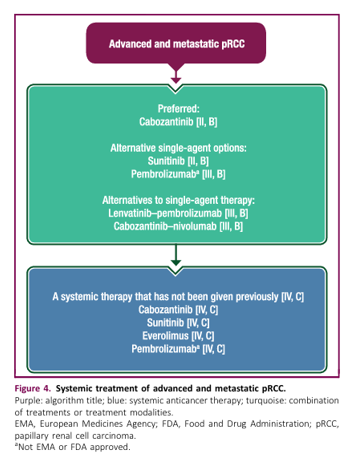
Hình 31. Lưu đồ lựa chọn phương pháp điều trị ung thư biểu mô tế bào thận thể nhú giai đoạn tiến triển và di căn.
- Cabozantinib là đơn trị liệu bước một được ưu tiên cho pRCC tiến triển, do đã chứng minh cải thiện sống không tiến triển (PFS) so với sunitinib, dù chưa chứng minh được lợi ích sống còn toàn bộ (OS).
- Các lựa chọn đơn trị liệu khác bao gồm sunitinib và pembrolizumab.
- Dữ liệu từ các nghiên cứu ngẫu nhiên quy mô nhỏ cho thấy savolitinib (một thuốc ức chế MET) có hoạt tính trong điều trị bước một pRCC có biến đổi gen MET.
Các thử nghiệm tiến cứu đơn nhánh đã đánh giá các phối hợp miễn dịch – điều trị đích, bao gồm: lenvatinib – pembrolizumab (n = 147), cabozantinib – nivolumab (n = 47), với tỷ lệ đáp ứng khách quan (ORR) lần lượt là 49% và 48%. Hồ sơ độc tính của các phối hợp này phù hợp với dự đoán, không ghi nhận tín hiệu an toàn bất thường.
Điều trị các dòng tiếp theo (Further-line treatment)
Hiện cũng thiếu dữ liệu mạnh cho điều trị bước hai trong pRCC.
Nguyên tắc thực hành là: Bất kỳ liệu pháp nhắm trúng đích hoặc miễn dịch nào đã được khuyến cáo ở bước một, nhưng chưa được sử dụng trước đó, đều có thể được cân nhắc một cách thận trọng ở các dòng sau.
Tuy nhiên, chưa có thử nghiệm ngẫu nhiên nào chứng minh lợi ích OS cho bất kỳ liệu pháp bước hai nào, và nguyên tắc điều trị theo chuỗi (sequencing therapy) chưa được chứng minh trong pRCC như ở ccRCC.
Ở một số bệnh nhân được chọn lọc kỹ, đặc biệt khi toàn trạng kém hoặc bệnh tiến triển chậm,chăm sóc giảm nhẹ tối ưu đơn thuần có thể là một lựa chọn phù hợp.
Điều trị ung thư biểu mô tế bào thận tiến triển và di căn không phải tế bào sáng, không phải thể nhú (non-clear-cell, non-papillary RCC)
Hiện nay, còn thiếu rất nhiều dữ liệu mạnh để hướng dẫn điều trị cho các thể RCC không phải tế bào sáng và không phải thể nhú. Các dữ liệu hiện có chủ yếu đến từ các nghiên cứu tiến cứu quy mô nhỏ và phân tích dưới nhóm của các thử nghiệm lớn hơn.
Phẫu thuật
Phẫu thuật có thể được sử dụng ở một số bệnh nhân bệnh tiến triển thuộc nhóm nguy cơ trung gian. Tuy nhiên, không có bằng chứng hiện tại đủ mạnh để ủng hộ phẫu thuật như một khuyến cáo chuẩn trong nhóm bệnh này.
Điều trị toàn thân (Systemic therapy)
Một thuật toán điều trị toàn thân cho RCC không phải tế bào sáng, không phải thể nhú tiến triển và di căn được trình bày trong Hình 5.
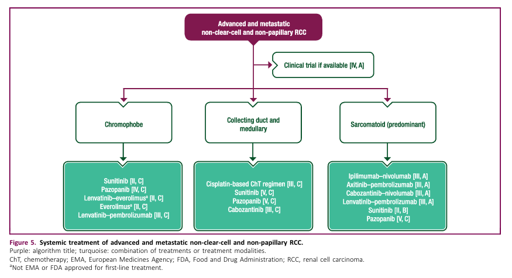
Hình 32. Lưu đồ lựa chọn phương pháp điều trị cho ung thư biểu mô tế bào thận không phải tế bào sáng hoặc thể nhú
- Sunitinib đã được chứng minh có hoạt tính sinh học trong các thể không phải tế bào sáng, với cải thiện sống không tiến triển (PFS) so với everolimus, từ đó ủng hộ vai trò của các thuốc ức chế tyrosine kinase (TKI) trong các phân nhóm hiếm này.
- Các phối hợp nhắm PD-1 được xem là tiêu chuẩn điều trị (standard of care) ở bệnh nhân có biệt hóa sarcomatoid, do hiệu quả vượt trội đã được ghi nhận.
- Ở một số bệnh nhân ung thư thận thể kỵ sắc (chromophobe RCC), thuốc ức chế mTOR có thể mang lại lợi ích. Điều này dựa trên cơ sở sinh học rằng đột biến nhiễm sắc thể 17 có thể dẫn đến mất gen FLCN, từ đó hoạt hóa quá mức con đường mTOR.
Các thể mô học đặc biệt
Các dòng điều trị tiếp theo
Sau điều trị bước một, không thể đưa ra khuyến cáo rõ ràng cho các dòng điều trị tiếp theo, do thiếu dữ liệu lâm sàng phù hợp.
Tổng hợp các khuyến cáo của ESMO năm 2024 về điều trị ung thư biểu mô tế bào thận tiến triển hay di căn xa
Vai trò của phẫu thuật và điều trị tại chỗ
- Cắt thận giảm khối u (cytoreductive nephrectomy, CN) nhìn chung nên tránh trong RCC tiến triển. CN chỉ nên được cân nhắc ở một số bệnh nhân được chọn lọc, thuộc nhóm nguy cơ thuận lợi hoặc trung gian, sau khi hội chẩn đa chuyên khoa (MDT). (Mức bằng chứng I, mức khuyến cáo B)
- CN trì hoãn (deferred CN) là một lựa chọn ở những bệnh nhân đạt đáp ứng gần hoàn toàn và bền vững tại các vị trí di căn sau điều trị toàn thân, sau MDT. (II, B)
- Lựa chọn bệnh nhân để áp dụng điều trị tại chỗ hoặc theo dõi trong bối cảnh di căn phải được thảo luận trong MDT. Mặc dù chưa có dữ liệu xác định chính xác quần thể phù hợp, cả hai chiến lược này nên tránh ở bệnh nhân có gánh nặng di căn cao, tái phát sớm, hoặc bệnh diễn tiến ác tính. (III, B)
- Cắt bỏ di căn (metastasectomy) không được khuyến cáo thường quy trong vòng 1 năm sau cắt thận. Tuy nhiên, ở bệnh nhân di căn số lượng ít đã được cắt bỏ hoàn toàn (M1 và NED), pembrolizumab bổ trợ có thể được chỉ định. (II, B; ESMO-MCBSv1.1: A)
Điều trị bước một cho ccRCC tiến triển và di căn
- Lenvatinib – pembrolizumab, axitinib – pembrolizumab hoặc cabozantinib – nivolumab được khuyến cáo là điều trị bước một cho ccRCC tiến triển, không phụ thuộc nhóm nguy cơ IMDC. Không có phối hợp PD-1 – VEGFR TKI nào được xem là ưu tiên hơn, và không khuyến cáo so sánh gián tiếp giữa các thử nghiệm. (I, A; ESMO-MCBSv1.1: 4, 4 và 1)
- Ipilimumab– nivolumab được khuyến cáo cho bệnh nhân IMDC nguy cơ trung gian và xấu, và là một lựa chọn ở bệnh nhân nguy cơ thuận lợi. (Trung gian/xấu: I, A; MCBS 4 —Thuận lợi: I, C)
- Axitinib– toripalimab là một lựa chọn cho bệnh nhân nguy cơ trung gian hoặc xấu. (I,C; chưa được EMA/FDA phê duyệt)
- Sunitinib, pazopanib và tivozanib là các lựa chọn thay thế cho phối hợp nhắm PD-1 ở bệnh nhân IMDC nguy cơ thuận lợi, do chưa có bằng chứng rõ ràng cho thấy phối hợpPD-1 vượt trội hơn sunitinib. (Sunitinib, pazopanib: I, C; tivozanib: II, C)
- Khi liệu pháp miễn dịch (ICI) chống chỉ định hoặc không sẵn có, sunitinib, pazopanib và tivozanib là các lựa chọn thay thế. Cabozantinib cũng là một lựa chọn cho bệnh nhân IMDC nguy cơ trung gian và xấu không thể dùng PD-1 bước một.(II, A)
- Axitinib – avelumab không cho thấy lợi ích OS so với sunitinib, do đó không được khuyến cáo. (I, D; ESMO-MCBS v1.1: 3)
- Theo dõi chủ động có thể là một chiến lược thay thế ở một nhóm nhỏ, chưa xác định rõ, bệnh nhân nguy cơ thuận lợi. (III, C)
- Ngừng ICI nên được cân nhắc sau 2 năm điều trị. Ngắt quãng điều trị VEGFR TKI không làm giảm hiệu quả. (ICI: IV, B; VEGFR TKI: I, C)
Điều trị bước hai cho ccRCC tiến triển và di căn
- Điều trị theo chuỗi bằng VEGFR TKI sau thất bại liệu pháp PD-1 bước một là tiêu chuẩn điều trị. Cabozantinib là lựa chọn ưu tiên.
(I, B; Cabozantinib: II, B)
- Các lựa chọn khác gồm axitinib, lenvatinib – everolimus, pazopanib, sunitinib,tivozanib. (III, B)
- Ở bệnh nhân đã điều trị bước một bằng VEGFR TKI, nivolumab (nếu dùng được) và cabozantinib đều cho thấy lợi ích OS. (Nivolumab: I, A; MCBS 5 — Cabozantinib:I, A)
- Belzutifan là một lựa chọn thay thế sau thất bại phối hợp VEGFR–PD-1.
(III, B; FDA phê duyệt, chưa EMA)
Điều trị các dòng sau cho ccRCC
- Có thể khuyến cáo tiếp tục điều trị theo chuỗi VEGFR TKI hoặc belzutifan. (VEGFRTKI: III, B — Belzutifan: I, B)
- Belzutifan nên được ưu tiên hơn everolimus ở bệnh nhân đã điều trị nhiều dòng. (I, B)
- Everolimus vẫn là một lựa chọn, nhưng không phải ưu tiên. (II, C)
- Không khuyến cáo tiếp tục điều trị bằng PD-(L)1 sau khi đã thất bại với PD-1 bước một.(I, D)
Điều trị toàn thân cho pRCC tiến triển
- Cabozantinib là đơn trị liệu bước một được ưu tiên nếu không có xét nghiệm phân tử bổ sung. (II,B)
- Lenvatinib – pembrolizumab và cabozantinib – nivolumab cho tỷ lệ đáp ứng cao, nhưng chưa chứng minh vượt trội so với đơn trị. (III, B)
- Các lựa chọn khác gồm sunitinib và pembrolizumab. Savolitinib hiện chưa được khuyến cáo. (II–III, B–D)
- Điều trị bước hai có thể dựa trên các thuốc chưa dùng trước đó; chăm sóc giảm nhẹ tối ưu (BSC) là một lựa chọn ở bệnh nhân chọn lọc. (IV, C)
Điều trị toàn thân cho RCC không phải tế bào sáng, không phải thể nhú
- Tham gia thử nghiệm lâm sàng được khuyến cáo. (IV, A)
- Sunitinib, pazopanib, everolimus, lenvatinib – everolimus, lenvatinib – pembrolizumab có thể được sử dụng cho chromophobe RCC.
- Hóa trị nền cisplatin được khuyến cáo cho collecting duct carcinoma và SMARCB1-deficient RCC; TKI là lựa chọn thay thế.
- Liệu pháp miễn dịch là ưu tiên trong RCC có biệt hóa sarcomatoid.
- Bevacizumab – erlotinib có thể dùng trong FH-deficient RCC.
Vai trò của xạ trị và thuốc bảo vệ xương
- Xạ trị định vị được khuyến cáo cho di căn não; xạ trị toàn não nên tránh do nguy cơ suy giảm nhận thức. (III, B / III, D)
- Zoledronicacid hoặc denosumab có thể được cân nhắc ở bệnh nhân di căn xương, sau khi đánh giá nguy cơ cá thể hóa. (IV, C)
Dự phòng
Một số yếu tố được coi là nguyên nhân hoặc tác nhân liên quan gây ung thư thận đã được xác định, bao gồm thuốc lá, béo phì,tăng huyết áp và yếu tố gia đình.
Để phòng bệnh ung thư thận: không hút thuốc lá, thuốc lào; tránh béo phì; loại trừ các nguyên nhân gây tăng huyết áp, điều trị tốt và kiểm soát bệnh tăng huyết áp; theo dõi, tầm soát định kỳ đối với các đối tượng trực hệ trong gia đình đã có người mắc ung thư thận.
Tiên lượng bệnh
Tiên lượng bệnh phụ thuộc vào giai đoạn, phân tầng nguy cơ, cũng như tuân thủ điều trị của người bệnh.
Bệnh có tiên lượng còn hạn chế do đa số trường hợp được phát hiện ở giai đoạn muộn. Phát hiện sớm mang lại tỷ lệ chữa khỏi bệnh cao.
Theo dõi định kỳ
Khi bệnh ổn định, có thể theo dõi bệnh nhân định kỳ.
Năm đầu: 3 tháng một lần; từ năm thứ hai trở đi: 6 tháng một lần.
Các xét nghiệm cần làm: công thức máu, sinh hóa máu, chụp tim phổi, siêu âm bụng, chụp CT scan bụng, xạ hình thận; chụp PET/CT khi cần.
Tài liệu tham khảo
1. Bộ Y tế (2021). Hướng dẫn chẩn đoán và điều trị một số bệnh ung bướu. Nhà xuất bản Y học.
2. Vincent T DeVita, Theodore S.Lawrence, Steven A. Rosenberg (2014). Cancer Principles and Practice of Oncology, 10th edition. Lippincott Ravell publishers. Philadelphia, UnitedStates.
3. Powles, T. et al., Renal cellcarcinoma: ESMO Clinical Practice Guideline for diagnosis, treatment andfollow-up, Annals of Oncology, Volume 35, Issue 8, 692 – 706
4. McCabe EM, Lee S, Rasmussen TP.Belzutifan (Welireg™) for von Hippel Lindau disease. Trends Pharmacol Sci. 2022Oct;43(10):882-883. doi: 10.1016/j.tips.2022.05.002. Epub 2022 Jun 9. PMID:35691787.
5. Ahrar, K., Matin, S. F., &Wallace, M. J. (n.d.). Thermal ablation of renal cell carcinoma. Radiology Key.Retrieved December 22, 2025, fromhttps://radiologykey.com/thermal-ablation-of-renal-cell-carcinoma/
6. Endolap Urology Clinic. (n.d.).Radical nephrectomy. Retrieved December 22, 2025, fromhttps://www.endolapurologyclinic.com/radical-nephrectomy/
7. Leibovich BC, Lohse CM, Cheville JC, et al. Renal CellCarcinoma with Inferior Vena Cava Extension: Can Classification Be Optimized toPredict Perioperative Outcomes? Kidney Cancer. 2020;4(2):111-115.doi:10.3233/KCA-190070
8. Gaballa, Salem & Hlaing, Kyaw & Mahler, Nathan &Moursy, Safa & Ahmed, Ameenjamal. (2020). A Rare Case of Immune-Mediated Primary Adrenal Insufficiency With Cytotoxic T-Lymphocyte Antigen-4 InhibitorIpilimumab in Metastatic Melanoma of Lung and Neck of Unknown Primary. Cureus.12. 10.7759/cureus.8602.
9. Eric A. Singer et al. Management of Metastatic Clear CellRenal Cell Carcinoma: ASCO Guideline Q&A. JCO Oncol Pract 19,127-131(2023). DOI:10.1200/OP.22.00660
10. Lee, CH., Motzer, R. The evolution of anti-angiogenictherapy for kidney cancer. Nat Rev Nephrol 13, 69–70 (2017). https://doi.org/10.1038/nrneph.2016.194
11. Popescu MC, Tretiakova M. Renal cell carcinoma overview. Pathology Outlines.com website.https://www.pathologyoutlines.com/topic/kidneytumormalignantrcc.html. AccessedDecember 21st, 2025.
12. Asadov D, Foster T, Weerakkody Y,et al. Renal cell carcinoma. Reference article, Radiopaedia.org (Accessed on 21Dec 2025). https://doi.org/10.53347/rID-7534
13. Rose, T. L., & Kim, W. Y.(2024). Renal Cell Carcinoma: A Review. JAMA, 332(12), 1001–1010.https://doi.org/10.1001/jama.2024.12848